






乳腺癌中表達NRP2的細胞是S-亞硝基化樞紐,可減輕輻射誘導的氧化應激
盡管新輔助化療極大的改善了乳腺癌患者的治療效果,但部分患者仍有殘留病灶。放射治療作為治療惡性腫瘤的主要手段之一,在影像技術和人工智能發展幫助下,可以為患者提供最佳的個性化治療。然而三陰性乳腺癌(Triple-negative breast cancer, TNBC)患者對放射治療具有抵抗性。在該研究中,研究者發現了一種蛋白質S-亞硝基化修飾介導的放療抵抗機制并設計了恢復放療敏感性的靶向抗體。該文章于2024年10月發表在《Journal of Clinical Investigation》,IF=13.3。
技術路線
研究結果
1.抑制VEGF/NRP2增強TNBC的放射敏感性
腫瘤干細胞(cancer stem cells, CSCs)在TNBC中富集,并且對電離輻射產生的ROS具有抵抗性。研究者已經發現VEGF通路通過單次跨膜蛋白NRP2維持CSC的功能,但是VEGF/NRP2通路在氧化應激緩沖方面的作用尚不清楚。
研究者通過對比人正常乳腺細胞和TNBC原代細胞單劑量放療后的基因表達譜,發現相比于對照組,22個編碼細胞表面蛋白的基因在放射治療的TNBC原代細胞中高表達。其中,NRP2與接受放療的TNBC患者不良預后相關(圖1A)。研究者發現在放療抵抗的TNBC細胞系中NRP2在細胞表面的表達量與輻射劑量相關(圖1B)。為研究NRP2表達是否是放療后存活的關鍵調節因素,研究者在輻射處理后的TNBC細胞中干擾了NRP2表達,并通過流式細胞術檢測凋亡細胞以及克隆形成實驗得到肯定的結果(圖1C和D)。研究者進一步使用NRP2 mAb(aNRP2-10)阻斷VEGF/NRP2結合,增加了TNBC細胞對輻射的敏感性(圖1E-G),表明VEGF/NRP2軸具有克服放療細胞毒性的潛力。
為評估數據的轉化相關性,研究者在體外實驗中模擬了臨床常規放療方案,發現放療抵抗的細胞中NRP2表達上調,使用aNRP2-10則增加了放療敏感性(圖1F)。研究者還使用了TNBC患者來源的類器官(PDOs)和異種移植物(PDXs)證明阻斷NRP2以增強放療敏感的有效性。免疫熒光和細胞活力實驗證明aNRP2-10處理使PDO對輻射敏感(圖1H)。依據NRP2細胞表面表達水平對PDX進行分選,研究者發現NRP2低表達細胞對放療更敏感,而aNRP2-10處理增加了NRP2高表達細胞對放療的敏感性(圖1I)。
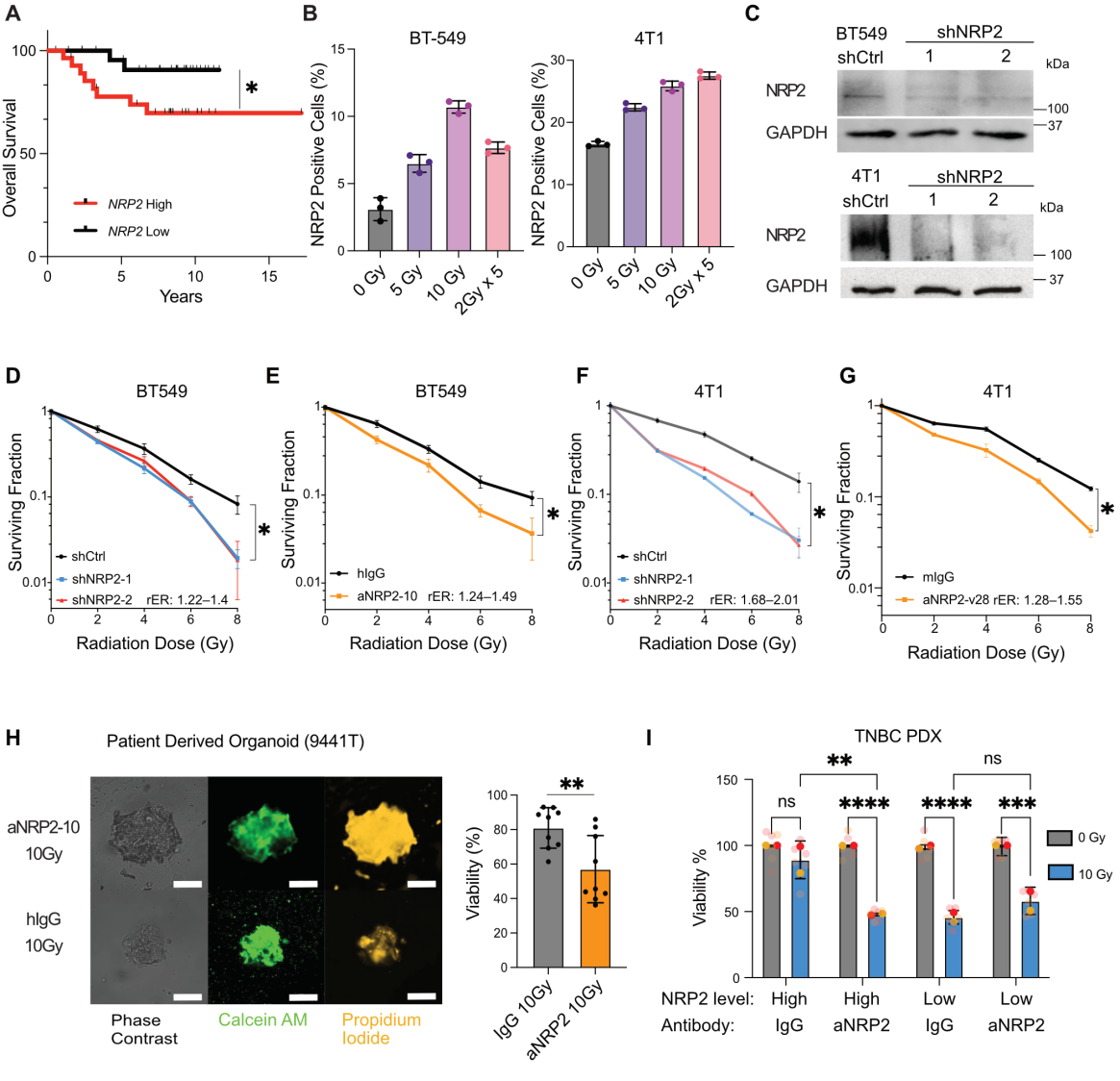
圖1 抑制VEGF/NRP2增強TNBC的放射敏感性
2.表達NRP2的細胞是NO信號傳導的樞紐
為研究NRP2促進放療抵抗的機制,研究者對NRP2低表達和NRP2高表達TNBC細胞的轉錄組進行了分析,發現一氧化氮合酶NOS2上調以及一氧化氮(NO)介導的信號轉導的相關基因富集(圖2A)。一氧化氮是一種能夠引發放射抗性的獨特生物活性信使。隨后,研究者通過免疫熒光檢測了患者組織中NO的替代標志物硝基酪氨酸和NRP2的表達,盡管硝基酪氨酸和NRP2的定位存在顯著的腫瘤內異質性,但Pearson共定位系數表明這兩個標記物之間存在很強的線性關系(圖2B)。研究者假定,NRP2表達的細胞通過調節NOS2表達,構成一個硝基化反應的核心樞紐。為證明VEGF/NRP2軸可以調節NOS2,研究者分別干擾了NRP2、VEGF-C的表達,以及使用aNRP2-10阻斷VEGF/NRP2結合,發現TNBC細胞中NOS2顯著下調(圖2C)。并且在NRP2敲降的細胞中,NO替代標志物亞硝酸鹽也減少(圖2D)。為評估NO在細胞內的擴散能力,研究者做了如下實驗:收集NRP2高表達細胞的培養基,添加DMSO或NO清除劑(c-PTIO),用于培養NRP2敲降的細胞。結果顯示NRP2高表達細胞的培養基增加了硝基酪氨酸含量,使用c-PTIO則消除了這種作用(圖2E)。
接下來,研究者從TCGA數據庫中選擇接受放射治療的癌癥患者,并根據其NRP2和NOS2 mRNA的表達進行分離。生存分析表明,NRP2和NOS2表達均高于中位的患者無病生存時間較短。為了驗證NRP2/NOS2軸在放療抵抗中的作用,使用化學抑制劑1400W或shRNAs抑制TNBC細胞中的NOS2活性,觀察到在寬范圍的輻射劑量下細胞存活率顯著降低。在NRP2敲降細胞中過表達NOS2則逆轉了放療抵抗的表型(圖2F)。重要的是,當用aNRP2-10處理時,NOS2過表達的TNBC細胞的放射敏感性沒有發生變化,這表明aNRP2-1治療后NOS2抑制對調節放射敏感性的必要性。

圖2 表達NRP2的細胞是NO信號傳導的樞紐
3.NRP2誘導NOS2表達以減輕輻射誘導的ROS
為進一步研究NRP2作用的硝基化反應介導放療抵抗的機制,研究者評估了減輕輻射誘導的關鍵因素:DNA損傷修復和氧化應激。DNA損傷修復的指標之一是追蹤照射后γ-H2AX焦點隨時間的衰減。相比于對照組,NRP2敲降細胞檢測到更多的γ-H2AX焦點,表明受到了更多的DNA損傷,或者其DNA損傷修復能力受到了影響(圖3A)。研究者進一步檢測了NRP2敲降細胞中ROS水平,ROS是引起DNA損傷積累的因素之一。使用ROS指示劑H2DCFDA,研究者發現輻射誘導NRP2敲降細胞和NRP2功能阻斷的細胞中ROS增加(圖3B和C)。在檢測DNA損傷的彗星實驗中,NRP2敲降細胞DNA損傷程度更高,而使用抗氧化劑N-乙酰半胱氨酸可以減輕NRP2敲降細胞DNA損傷,而對對照細胞沒有作用(圖3D)。以上實驗表明,VEGF/NRP2可以最大限度地減少輻射誘導的ROS積累,并減輕DNA損傷。為檢測VEGF/NRP2軸對ROS的緩沖作用是否依賴NOS2活性,使用1400W抑制NOS2功能,結果發現在NRP2敲降細胞中抑制NOS2,ROS水平沒有明顯變化,而對照組細胞抑制NOS2功能導致ROS水平上升(圖3E)。并且在NRP2敲降細胞中過表達NOS2可以減少輻照誘導的ROS水平(圖3F)。以上結果表明,NRP2/NOS2軸察省的NO可以減輕輻射誘導的氧化應激導致的DNA損傷。
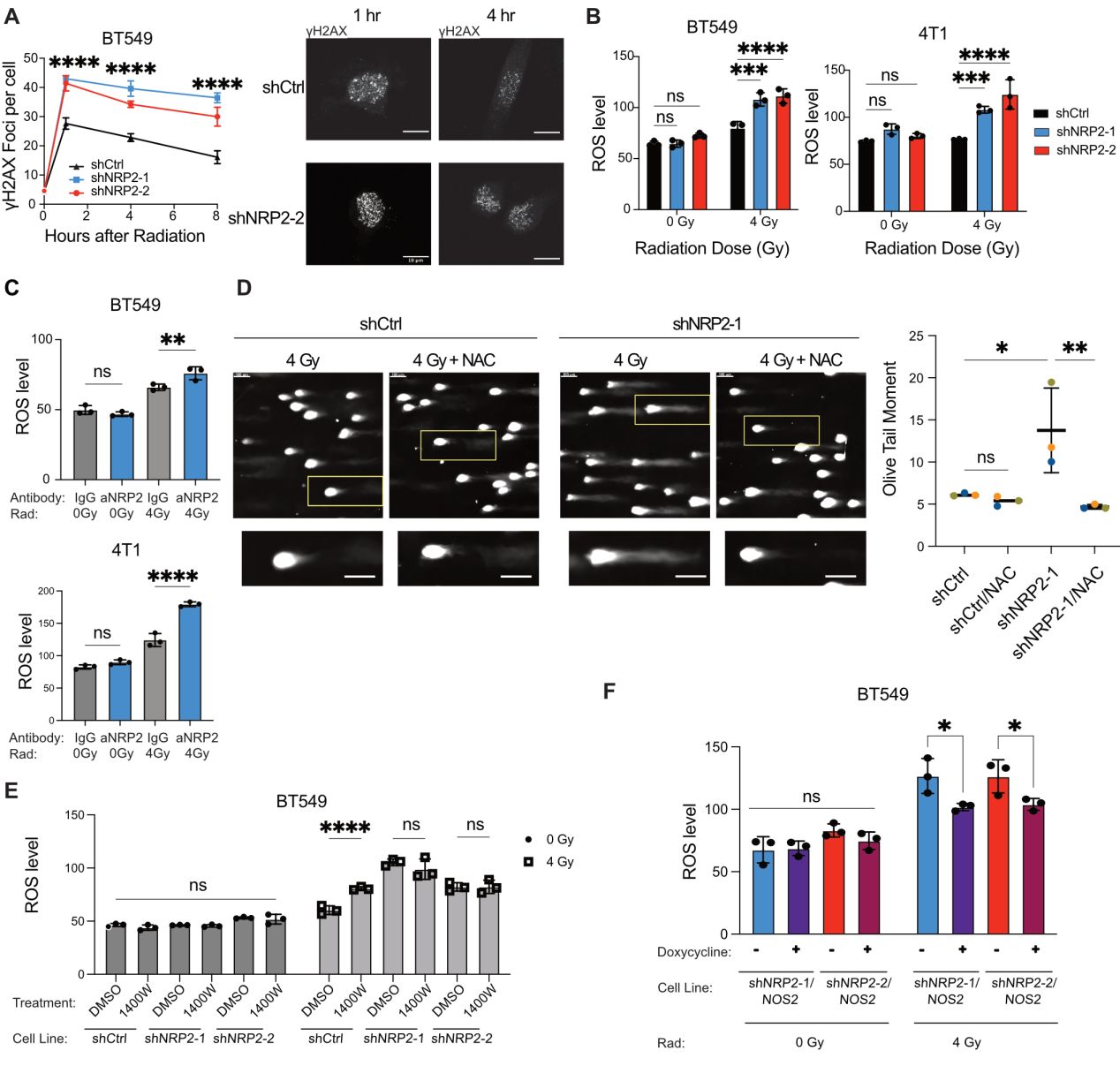
圖3 NRP2誘導NOS2表達以減輕輻射誘導的ROS
4.VEGF/NRP2通過Gli1調節NOS2轉錄
研究者過去研究發現,VEGF/NRP2下游信號通路誘導轉錄因子Gli1表達,在NRP2敲降和功能阻斷細胞中Gli1表達降低(圖4A和B)。并且,輻射增加了Gli1表達,但是在NRP2功能阻斷后Gli1表達降低(圖4C)。研究者假設NOS2是Gli1的靶基因,使用Gli1抑制劑GANT61處理TNBC細胞,發現NOS2 mRNA表達顯著降低(圖4D)。敲降Gli1得到了一致的實驗結果(圖4E)。而在NRP2敲降細胞中過表達Gli1則增加NOS2表達(圖4F)。研究者結合已發表的Gli1 ChiP-seq數據和ChIP-qPCR實驗,證明了Gli1可以與NOS2啟動子結合(圖4G)。為進一步證明Gil1驅動NOS2轉錄,研究者用CRISPR-Cas9技術敲除了Gil1與NOS2啟動子結合的轉錄調控元件,結果導致NOS2 mRNA表達減少(圖4H),并且這些細胞對輻照敏感(圖4I)。以上結果說明NOS2表達依賴于Gli1的轉錄調控。
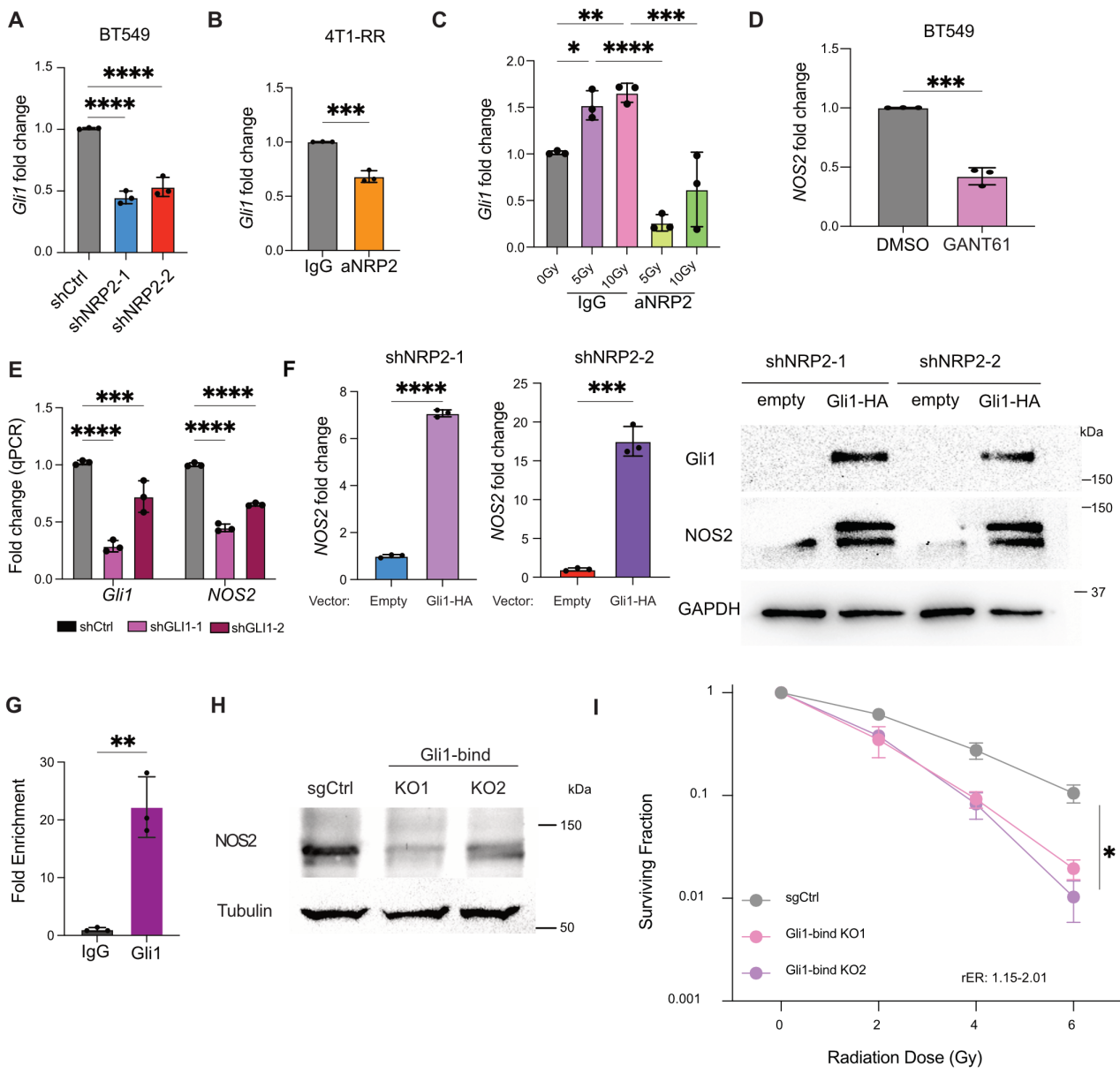
圖4 VEGF/NRP2通過Gli1調節NOS2轉錄
5.NFE2L2活性依賴于NRP2誘導的NOS2表達
研究者接下來通過分析放療敏感的TNBC細胞系和放療抵抗的TNBC細胞系的轉錄組數據,進一步探索下游機制。NOS2 mRNA在放療抵抗的TNBC細胞系中上調,并且NFE2L2/KEAP1通路在差異基因中富集。NFE2L2是抗氧化反應元件的關鍵調節因子,可以減輕癌癥細胞中的ROS水平,并受NO調節。研究者假設,VEGF/NRP2通過誘導基于KEAP1(Cullin 3的泛素連接酶的成分之一)的S-亞硝基化,促進NFE2L2定位到細胞核。免疫熒光證明NRP2敲降和功能阻斷細胞中,NFE2L2核定位減少(圖5A和B)。研究者借助檢測NFE2L2靶基因的表達證明NRP2敲降導致NFE2L2穩定性降低(圖5C)。為證明NFE2L2依賴NOS2發揮活性,研究者敲降NOS2表達或者抑制其功能,發現NFE2L2核定位減少取決于NOS2表達,而不是NRP2(圖5D)。
研究者發現使用NO清除劑c-PTIO會導致NFE2L2靶基因轉錄本減少;用對照組的培養液處理NRP2敲降細胞導致靶基因轉錄本增多,添加c-PTIO則會抵消這種效果。向NRP2敲降細胞中添加NO供體SNAP可以增加NFE2L2核定位(圖5E)。接下來,研究者對在NRP2表達的細胞中NFE2L2靶基因的表達取決于NO產生和隨后KEAP1的S-亞硝基化進行驗證。研究者通過生物素轉化和碘-TMT轉化檢測S-亞硝基化水平。NRP2敲降細胞中KEAP1的S-亞硝基化水平明顯降低(圖5F和G)。進一步,研究者構建了一個沒有KEAP1結合結構域的活性NFE2L2(caNFE2L2)質粒,轉入NRP2敲降細胞中。caNFE2L2的表達增加了輻射抵抗(圖5H)。研究者發現分級輻射僅能在對照細胞中誘導NFE2L2激活,而NRP2敲除細胞沒有這種反應(圖5J)。這一結果表明,腫瘤在放射治療期間依賴于表達NRP2的細胞來促進NFE2L2的激活,從而促進抵抗性。
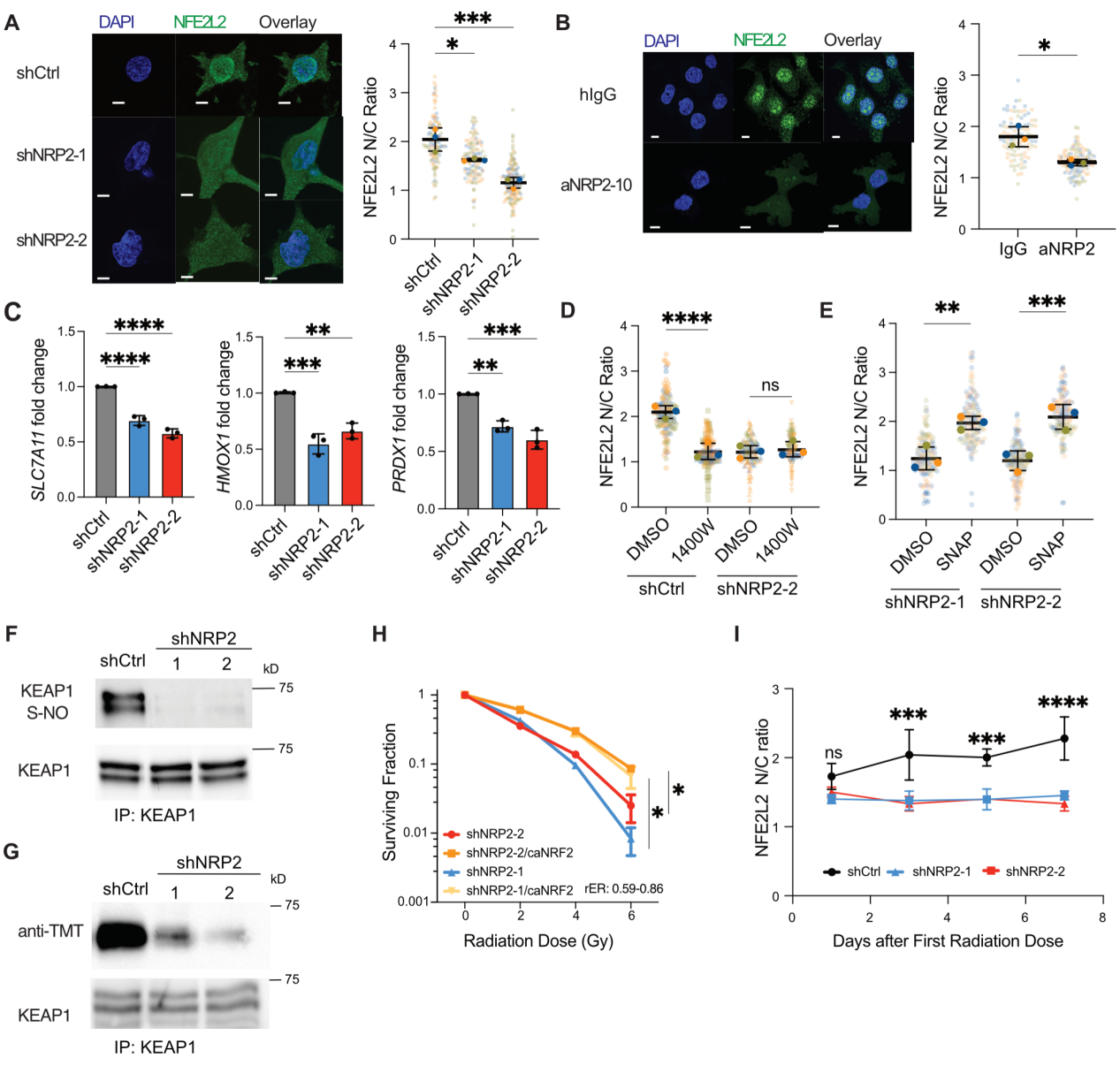
圖5 VEGF/NRP2通過Gli1調節NOS2轉錄

圖6 NFE2L2活性依賴于NRP2誘導的NOS2表達
6.單劑量或常規分次放療聯合VEGF/NRP2抑制可延緩腫瘤生長
為了探究VEGF/NRP2抑制增強體內放射敏感性的潛力,研究者首先使用小鼠4T1三陰性乳腺癌(TNBC)細胞和單劑量放療建立了同種異體移植模型。單次10Gy放療劑量與aNRP2-28治療的組合顯著抑制了腫瘤生長,與單獨任一治療相比(圖6A)。此外,聯合治療比其他治療方案增加了腫瘤中的壞死(圖6B)。使用γH2AX作為放射敏感性的可靠標記(37, 38),研究者觀察到與IgG對照相比,VEGF/NRP2功能阻斷抗體在放療后增加了γH2AX的表達(圖6C)。為了評估腫瘤的增殖能力,研究者分析了Ki-67的表達,并量化了有絲分裂細胞的數量。用aNRP2-28治療的腫瘤在Ki-67陽性細胞的百分比上顯著降低(圖6D)并且有絲分裂細胞數量減少,表明VEGF/NRP2功能阻斷抗體也限制了放療后的腫瘤增殖。研究者證實了之前的研究結果,即VEGF/NRP2抑制抑制了NOS2(圖6,E和F)和NFE2L2靶基因(圖6G)的表達。與對照組相比,放療的腫瘤中NOS2和NFE2L2靶基因的表達更高,而用aNRP2-28治療則減輕了這些通路的表達。然后,研究者分析了常規分割放療與aNRP2-28的組合效果。與單劑量放療實驗類似,VEGF/NRP2抑制與常規分割放療的組合減輕了腫瘤的生長潛力(圖7A),在腫瘤內增加了壞死(圖7B),并且增加了γH2AX的保留(圖7C)與單獨IgG與傳統分割放療組相比。此外,aNRP2-28和放療的聯合治療降低了NOS2(圖7D)和NFE2L2靶基因(圖7E)的表達,與單獨傳統分割放療組相比。關于aNRP2的作用和安全性,小鼠體重或正常乳腺實質的組織學變化沒有顯著變化。

圖7 單劑量或常規分次放療聯合VEGF/NRP2抑制可延緩腫瘤生長
7.aNRP2分次放療促進腫瘤消退
放療技術的進步和幾項臨床試驗推動了在早期、低風險乳腺癌中使用低分割放療。標準治療通常需要25次分割的50Gy,而低分割減少了5-15次分割的數量,每次分割的輻射劑量更高。在這個實驗中,研究者使用了一種侵襲性的TNBC PDX模型。當腫瘤達到適當的大小時,研究者開始按照圖8A中概述的進行放療和抗體治療。單獨的分割放療或單獨的aNRP2-10對腫瘤生長沒有影響,這與TNBC對放療的已知不敏感性一致。然而,聯合治療導致顯著的腫瘤退縮(圖8B),降低了腫瘤重量并增加了壞死(圖8C)。研究者使用相同的實驗方法與4T1移植模型,也導致與單獨分割放療相比,聯合治療組腫瘤退縮、腫瘤重量減輕和壞死增加。此外,與其它組相比,聯合治療組的腫瘤中檢測到更高水平的γH2AX(圖8D)。研究者觀察到與單獨放療相比,聯合治療組的腫瘤中NOS2和NFE2L2靶基因的表達顯著降低(圖8E)。最后,aNRP2與低分割放療的聯合治療對小鼠體重沒有影響,與單獨低分割組相比,在PDX或4T1模型中均無影響。
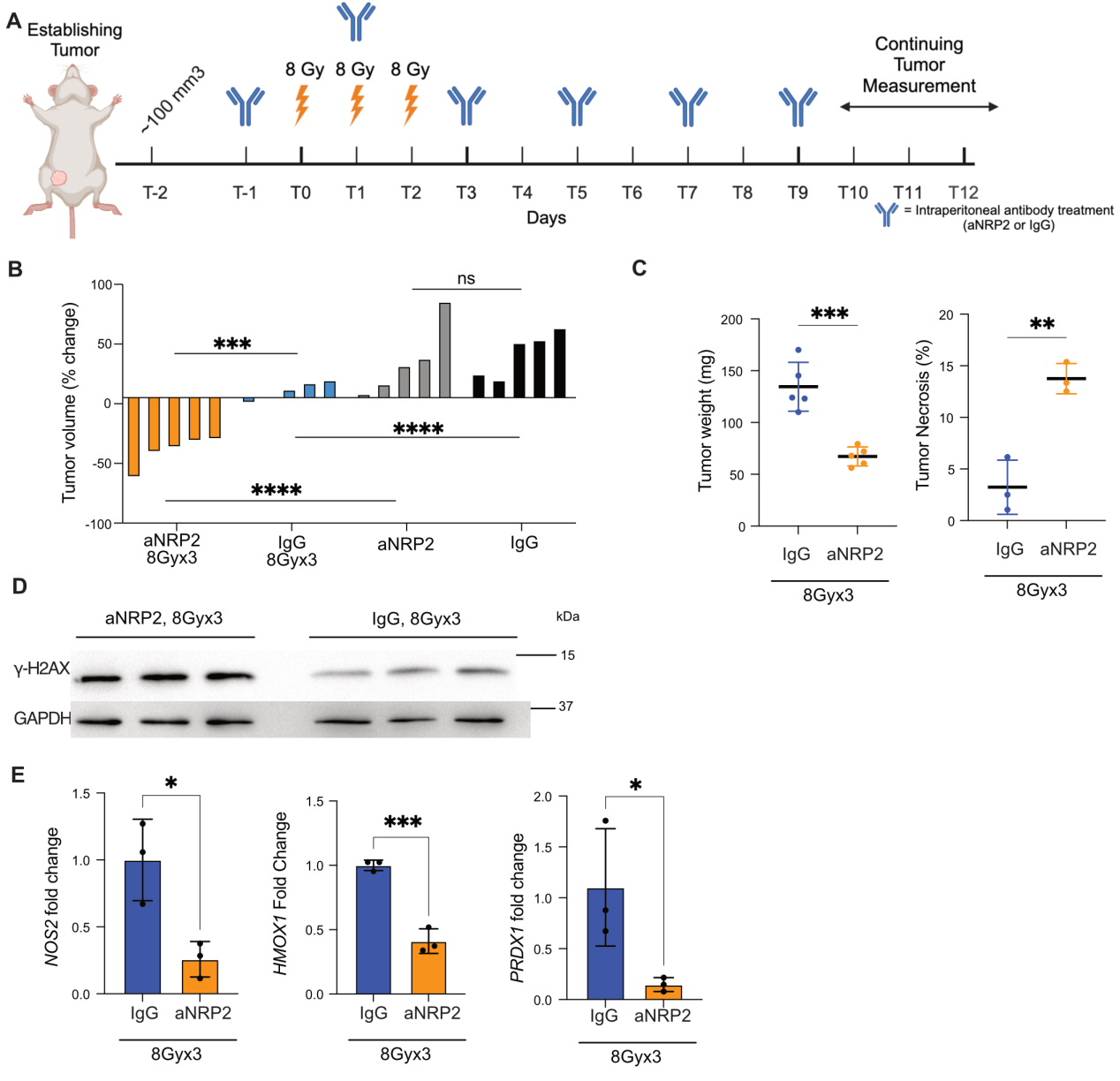
圖8 aNRP2分次放療促進腫瘤消退
結果
這項研究揭示了癌癥中的一種信號通路,可用于提高腫瘤對放射治療的敏感性。具體而言,TNBC中具有依賴于VEGF/NRP2信號傳導的干細胞特性的細胞亞群作為局部產生NO的樞紐,促進KEAP1等效應蛋白的S-亞硝基化,使NFE2L2介導的抗氧化基因得以表達。因此,腫瘤細胞可以緩沖輻射誘導的ROS的積累,并減輕其下游效應,包括DNA損傷和細胞死亡。使用功能阻斷的NRP2單克隆抗體來阻斷這些細胞的亞硝化能力,恢復腫瘤細胞對放射治療敏感性。
實驗方法
基因表達譜分析、轉錄因子結合位點分析、RNA測序、免疫熒光和共定位分析、ChIP-qPCR實驗、CRISPR-Cas9基因編輯、S-亞硝基化檢測、細胞培養與輻射處理、克隆形成實驗、細胞活力檢測、流式細胞術、ROS檢測、堿性彗星實驗、NO濃度檢測、同種異體移植模型、患者來源的異種移植物模型
參考文獻
Kumar A, Goel H, Wisniewski C, et al. Neuropilin-2 expressing cells in breast cancer are S-nitrosylation hubs that mitigate radiation-induced oxidative stress. J Clin Invest. Published online October 1, 2024. doi:10.1172/JCI18136