






siRNA修飾的lncRNA納米藥物通過抑制心肌內皮細胞鐵死亡治療心肌肥厚
心臟微血管功能障礙與心肌肥厚相關,最終可導致心力衰竭。長鏈非編碼RNA (lncRNAs)的異常調節被認為是心肌肥厚的關鍵機制之一。本文發表在《Molecular Therapy-Nucleic Acids》IF:8.886三月刊上。然而,lncRNA在心臟微血管功能障礙中的潛在作用和潛在機制尚未明確闡明。本研究結果證實心臟微血管功能障礙與心肌肥厚和心肌肥厚時心肌微血管內皮細胞(CMECs)鐵死亡有關。
技術路線:

主要實驗結果:
1、心肌肥厚時受損的心肌微血管發生鐵死亡
不同形式的死亡,如凋亡,自噬和壞死性凋亡都已被證明和心肌肥厚相關。這些結果表明不同形式的死亡方式可以共同作用于心肌肥厚,那么一些新的細胞死亡方式可能會被忽略。鐵死亡是一種鐵依賴的,非凋亡性細胞死亡,部分是由致死性脂質ROS積累介導的。此外,鐵死亡參與心肌損傷。基于以上作者猜想鐵死亡可能也參與了心肌肥厚。檢測結果發現鐵死亡相關蛋白ferrostatin-1 (Ferr-1)減少了心肌肥厚模型組心臟中脂質過氧化的增加,通過檢測MDA和ptgs2水平(圖1A-B)。并且,Ferr-1處理減少了模型組心臟H2AX的表達(圖1C-D)。此外,電子顯微鏡下觀察到心肌微血管內皮線粒體在AAC處理大鼠中顯著變形和萎縮,這是鐵死亡發生的典型標志,而這些現象在Ferr-1處理后顯著減少(圖1E)。明膠-墨水染色顯示Ferr-1顯著增加了心臟微血管密度(圖1F),并可以改善心肌微血管功能,表現為p-eNOS蛋白,NO含量和cGMP增加(圖1G-1J)。進一步研究發現,Ferr-1處理減少了ROS、脂質過氧化、ptgs2表達的增加(圖1K-1M),并增加了AngⅡ誘導的CMEC細胞活力(圖1N-1O),p-eNOS的表達(圖1R-1S),并增加了AngⅡ誘導的CMEC細胞中的NO含量。這些結果表明鐵死亡發生在心肌微血管損傷過程中,提示Ferr-1對肥厚性心臟的保護作用可能通過抑制鐵死亡來改善CMEC功能。

圖1抑制鐵死亡改善CMECs的功能
2、心肌肥厚心臟中MMP9/TIMP1比值失衡導致CMECs功能障礙
MMP9/TIMP1的失衡參與多種疾病的發生發展,作者的前期研究也發現心肌肥厚大鼠心臟和CMEC細胞中均存在MMP9/TIMP1失衡。在這里,作者證明TIMP1沉默導致MMP9表達顯著增加(圖2A-D),以及增加AngⅡ誘導的CMEC細胞活力(圖2E-F)。為探討MMP9/TIMP1失衡對CMECs功能的影響,采用免疫熒光和western blot檢測PECAM-1的表達,結果發現在AngⅡ處理的CMECs中,PECAM-1的表達明顯缺失,而TIMP1的沉默顯著提高了PECAM-1的表達(圖2G-2I)。此外,Ang II抑制細胞遷移和管形成能力,p-eNOS表達和NO含量,而TIMP1沉默顯著反轉這些作用(圖2J-N)。這些結果表明MMP9/TIMP1失衡導致CMECs功能障礙。
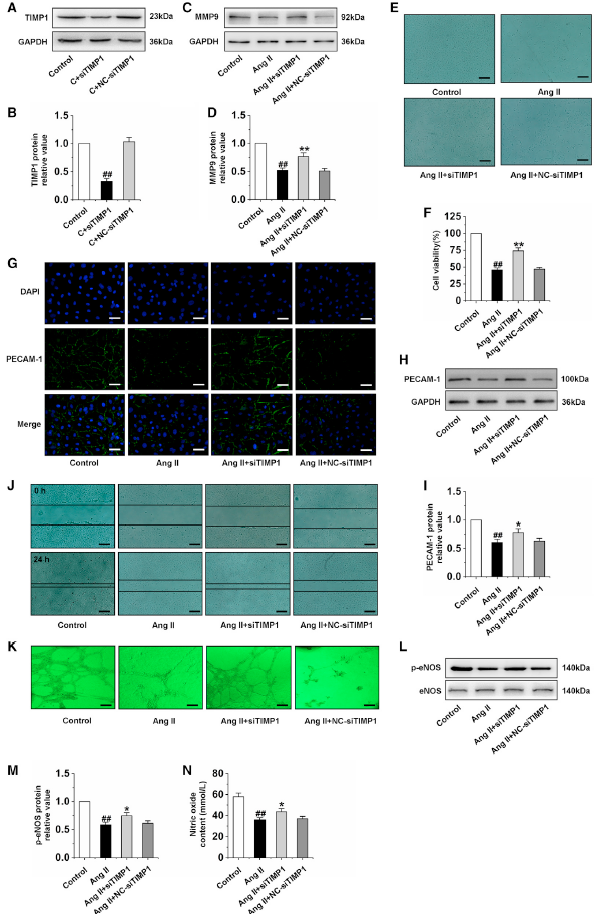
圖2沉默TIMP1改善AngⅡ誘導的CMECs功能異常
3、MMP9/TIMP1比值失衡誘導CMECs鐵死亡通過激活TFR-1
隨后探究TIMP1沉默保護CMEC功能的機制。作者檢測了鐵死亡相關蛋白的表達,包括SLC7A11,GPx4,TFR-1,Fpn1,結果表明沉默TIMP1顯著抑制AngⅡ誘導的TFR-1表達的增加,但對SLC7A11,GPx4,Fpn1蛋白表達沒有影響 (圖3A-B)。進一步研究表明,沉默TIMP1顯著降低了鐵離子水平,ROS和MDA的含量,以及ptgs2 mRNA的表達(圖3C-3F)。圖3G表明沉默TIMP1可以抑制CMECs細胞的死亡。提示MMP9/TIMP1比值的降低通過激活TFR-1導致CMEC鐵死亡。

圖3沉默TIMP1抑制CMEC鐵死亡通過下調TFR-1
4、miR-30b-5p抑制AngⅡ誘導的CMECs的鐵死亡
通過在線軟件發現miR-30b-5p與TIMP1的3’UTR區域有互補配對序列(圖4A)。熒光素酶實驗揭示miR-30b-5p能夠抑制野生型(WT) TIMP1的熒光素酶活性,而TIMP1 3’UTR的突變形式對miR-30b-5p的反應較弱(圖4B)。心肌肥厚大鼠心肌組織和Ang II誘導的CMECs中miR-30b-5p降低(圖4C-4D)。下一步將miR-30b-5p mimic和AMO-miR-30b-5p轉染至CMECs,檢測miR-30b-5p對CMECs鐵死亡反應的影響(圖4E)。在Ang II-induced CMECs,過表達miR-30b-5p導致TIMP1和TFR-1蛋白表達,ROS和MDA含量,鐵離子濃度,ptgs2的mRNA水平顯著降低,并顯著提高細胞生存能力和MMP9的蛋白表達,而這些效果能被AMO-miR-30b-5p逆轉(圖4F-M)。表明過表達miR-30b-5p可顯著抑制暴露于Ang II的CMECs的鐵死亡。然而,AMO-miR-30b-5p抑制了miR-30b-5p對CMECs的保護作用(圖4N)。
隨后,lncRNA的ceRNA機制調控引miR-30b-5p起了作者的興趣。作者發現有11個lncRNA與miR-30b-5p共享一個高度保守的結合位點。檢測了這11個lncRNA在心肌肥厚大鼠心肌組織中的表達變化,結果顯示lncRNA AC115159.2、lncRNA AABR07017145.1 (lncRNA AAB)和lncRNA AABR07030334.1的mRNA表達顯著上調(圖4O)。qRT-PCR檢測這三種lncRNA在CMECs中對Ang II處理的響應,發現在這些lncRNA中,只有lncRNA AAB顯著升高(圖4P)。圖4Q展示了lncRNA AAB和miR-30b-5p之間的結合位點。熒光素酶實驗顯示,miR-30b-5p可抑制TIMP1的熒光素酶活性,但該抑制可被lncRNA AAB逆轉,AAB-mut突變形式對TIMP1的熒光素酶活性影響較小(圖4R)。這些結果表明lncRNA AAB通過與miR-30b-5p相互作用,進而調控CMECs的鐵死亡。
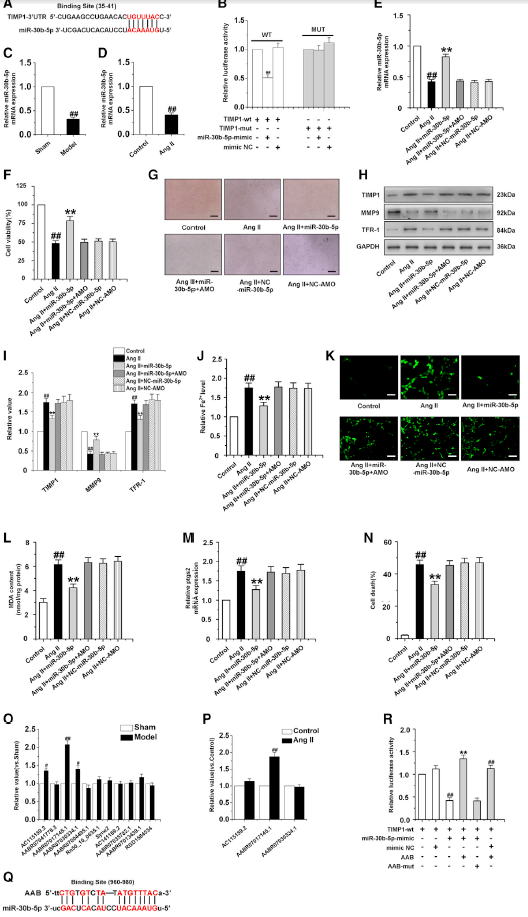
圖4 miR-30b-5p抑制AngⅡ誘導的CMECs的鐵死亡
5、納米復合物的制備、表征和細胞攝取
因此,如果能夠通過納米復合物有效地傳遞lncRNA AAB的小干擾RNA (siRNA),就有可能抑制CMECs的鐵死亡。接下來,作者展示了納米復合物的制備和表征。如圖5A所示,中性粒細胞膜(NM) + si-AAB +單聚二氧化硅納米復合物(MSN)清楚地表現出真核細胞樣結構,最外層為淺灰色的軟層,厚度為10 nm,與活細胞膜的厚度一致。所得到的MSNs為均勻的納米球,平均粒徑約為100 nm,如掃描電鏡圖像所示(圖5B-5C)。將孔徑調整到適合siRNA負載的大小,表面積保證了siRNA的高負載能力。此外,MSNs可進行生物降解當在胎牛血清中孵育24 h,這對體內藥物有效的釋放和減少機體免疫副作用和積累具有重要意義。通過zeta電位的測定來研究MSN的表面修飾和電荷變化。當MSN上載siRNA時,zeta電位下降到-14.6±1.06 mV,這與siRNA的負電荷有關(圖5D)。為了進一步證實MSNs的介孔結構,檢測了MSNs納米粒子的比表面積和孔徑。值分別為519 m2 g?1和2.4 nm左右排列良好的介孔(圖5E插圖),可以滿足后續siRNA的加載。x射線衍射(XRD)圖顯示,由于MSNs是無定型的,所以MSNs沒有明顯的峰值,但在2θ(22°)處有一個寬衍射峰(圖5F)。通過SDS-PAGE凝膠電泳,觀察到細胞膜和NM + si-AAB + MSN樣品中LFA-1和β2整合素的條帶(圖5G)。LFA-1和β2整合素的存在,保證了NM + si-AAB +MSN對損傷CMECs的有效靶向作用。
為了證明Ang II可以誘導CMECs損傷和炎癥,通過western blot檢測了ICAM-1的水平(圖5H-5I)。為了證明NM + si-AAB + MSN對CMECs的特異性識別和靶向傳遞,使用Ang II誘導CMs和CMECs損傷,然后和NM + si-AAB + MSN共孵育,并進行染色。在CM中觀察到的FITC信號較弱,表明對NM + si-AAB + MSN的攝取較少,而在CMEM中,FITC信號增強了3.8倍(圖5J-5K)。此外,細胞攝取si-AAB + MSN和NM + si-AAB + MSN的結果經過流式分析,發現NM + si-AAB + MSN組的攝取信號顯著增加(圖5L-5M)。以上表明NM + si-AAB + MSN可以被CMEM特異性獲取。
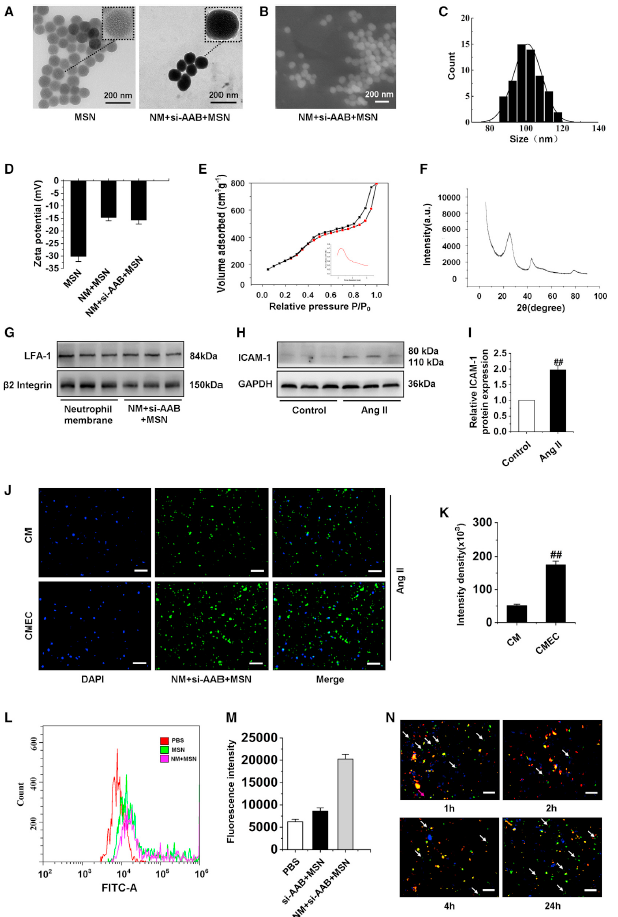
圖5納米復合物的準備和鑒定
6、NM + si-AAB + MSN抑制CMEM鐵死亡通過上調miR-20b-5p
圖6A展示了細胞內化和納米復合物細胞內結構解構,包括靶向識別、細胞內化、胞內細胞膜分離,和NM + si-AAB + MSN的降解以及siRNA釋放。在這種良好的減少反應下,NM + si-AAB + MSN可以有效地沉默CMECs中lncRNA AAB的表達,當siRNA劑量為30 nM時可以抑制超過70%的lncRNA AAB的表達(圖6B)。圖6C和圖6D顯示,和AngⅡ組相比,si-AAB + MSN和NM + si-AAB + MSN組的CMEM細胞活力顯著增加。圖6E表明AngⅡ誘導導致CMEM細胞的PECAM-1不完全染色,而si-AAB + MSN和NM + si-AAB + MSN組的外周染色均勻。進一步研究發現si-AAB + MSN和NM + si-AAB + MSN顯著上調miR-30b-5p的表達和MMP9的蛋白表達,下調TIMP1和TFR-1的蛋白表達(圖6F-6H)。與Ang II組相比,si-AAB組的鐵離子、ROS、MDA、ptgs2水平和細胞死亡幾乎沒有變化,而si-AAB +MSN和NM + si-AAB +MSN組的鐵離子、ROS、MDA、ptgs2水平和細胞死亡明顯降低,且NM + si-AAB +MSN組最低,表明NM + si-AAB +MSN可以有效抑制鐵死亡(圖6I-6M)。這些結果表明,NM + si-AAB + MSN通過上調miR-30b-5p對Ang II暴露的CMECs的鐵死亡抑制作用最有效。
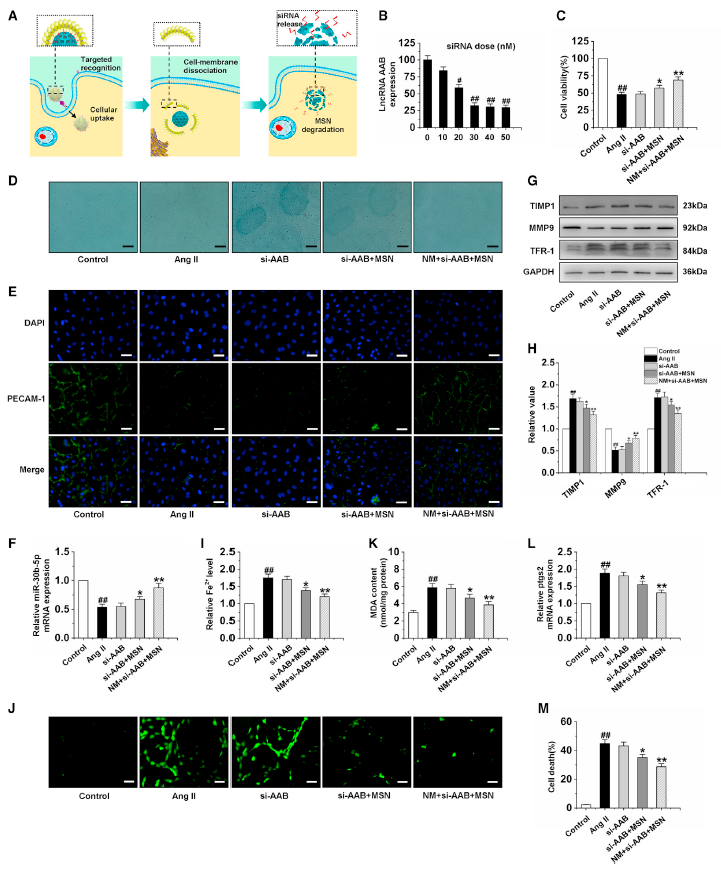
圖6通過NM+si-AAB+MSN沉默lncRNA AAB反轉CMEC的鐵死亡
7、NM+si-AAB+MSN修復心肌肥厚的心肌微血管內皮的損傷
在AAC處理建立心肌肥厚動物模型后,將心肌肥厚大鼠隨機分為4組。24 h后,心肌肥厚大鼠分別經尾靜脈注射PBS、si-AAB、si-AAB +MSN、NM + si-AAB +MSN,每2 d注射1次,連續4周(圖7A)。為了確保NM + si-AAB + MSN始終能夠靶向心臟,在AAC后的第3天、第9天和第27天測定了心肌組織中ICAM-1的蛋白水平。結果顯示,ICAM-1在這些不同時間點的蛋白表達明顯高于假手術組(圖7B和7C)。然后,通過定量MSN熒光信號的生物成像研究了納米復合物在心臟的分布。si-AAB + MSN組心臟出現輕微熒光信號;在NM + si-AAB + MSN組中,心臟中觀察到明顯信號,表明納米復合物對受損的內皮細胞具有良好的靶向特異性(圖7D)。觀察NM + si-AAB +MSN對肥厚心臟微血管鐵死亡的體內治療作用。電鏡觀察結果顯示,PBS處理模型組CMEC細胞線粒體萎縮(圖7E)。si-AAB組和si-AAB +MSN組的鐵離子水平變化不明顯,而NM + si-AAB +MSN組的鐵離子水平明顯下降,說明NM + si-AAB +MSN對心肌微血管具有良好的抗鐵死亡作用。此外,NM + si-AAB + MSN可增加心肌微血管密度(圖7F),并在恢復LVEF和LVFS方面表現較好(圖7G-7I)。腦室組織病理學顯示與PBS處理模型組相比,只有NM + si-AAB + MSN組產生的截面積更小,表現出明顯的心臟形態保護作用(圖7J和7K)。與模型組相比,si-AAB和si-AAB +MSN組BNP和β-MHC蛋白水平變化不大,而NM + si-AAB +MSN組BNP和β-MHC蛋白水平顯著降低,表明NM + si-AAB +MSN在體內能有效抑制心肌肥厚(圖7L-7M)。以上表明,M+si-AAB+MSN能在體內修復心肌肥厚的心肌微血管內皮的損傷。
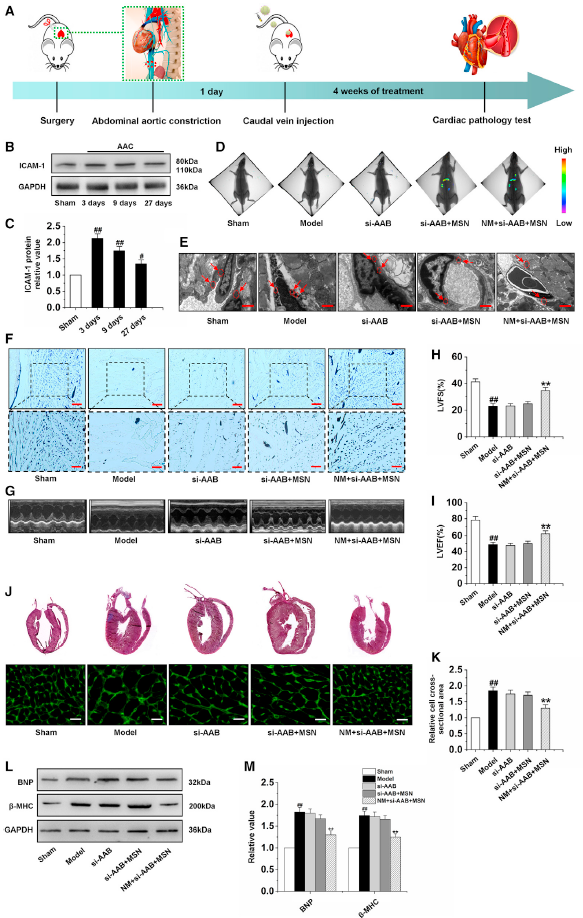
圖7通過NM+si-AAB+MSN沉默lncRNA AAB抑制心肌肥厚
總之,如圖8所示,本研究通過合理設計納米復合物,實現了si-AAB高效加載和細胞膜覆蓋,從而有效地履行其對心肌肥厚內皮損傷的精確修復功能。
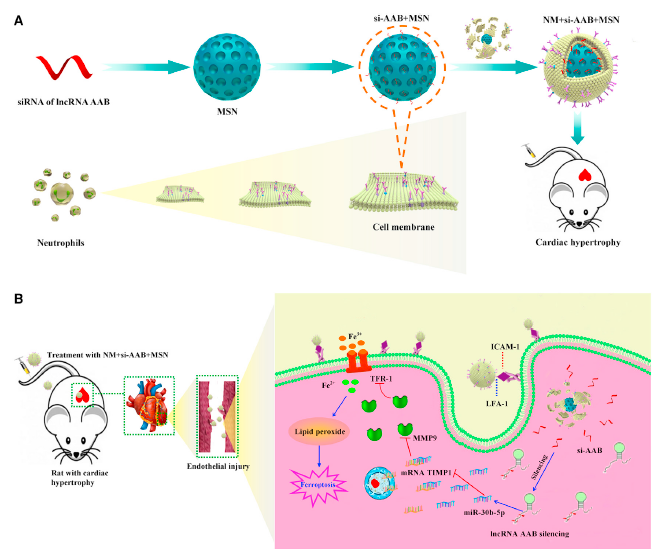
圖8 NM+si-AAB+MSN通過抑制CMEC鐵死亡調節心肌肥厚的機制模型圖
參考文獻:
Shi Pilong., Li Minghui., Song Chao., Qi Hanping., Ba Lina., Cao Yonggang., Zhang Meitian., Xie Yawen., Ren Jing., Wu Jiabi., Ren Ping., Sun Hongli.(2022). AABR07017145.1Neutrophil-like cell membrane-coated siRNA of lncRNA therapy for cardiac hypertrophy via inhibiting ferroptosis of CMECs. Mol Ther Nucleic Acids, 27(undefined), 16-36. doi:10.1016/j.omtn.2021.10.024