






小膠質細胞中YY1乳酸化通過轉錄激活介導的FGF2上調促進血管生成
眼部新生血管形成是導致失明的主要原因。視網膜小膠質細胞與缺氧誘導的血管生成和血管病變有關,但其潛在機制尚不完全清楚。乳酸化是一種新型的乳酸衍生翻譯后修飾,在多個細胞過程中起關鍵作用。由于缺血性視網膜病變中的缺氧是視網膜新生血管形成的誘發因素,因此乳酸化很可能參與這一過程。該研究旨在探討乳酸化在視網膜新生血管形成中的作用,并確定視網膜新生血管疾病的新治療靶點。集落刺激因子1受體(CSF1R)抑制劑對小膠質細胞耗竭PLX3397抑制氧誘導的視網膜病變中的視網膜新生血管形成。缺氧可增加小膠質細胞中的乳酸化并加速FGF2的表達,從而促進視網膜新生血管形成。作者確定了67個蛋白質的77個位點,在缺氧下乳酸增加的背景下乳酸化增加。結果表明,轉錄因子非組蛋白陰陽-1(YY1)在賴氨酸183(K183)位點乳酸化,賴氨酸183受p300調控。高乳酸化的YY1直接增強FGF2轉錄并促進血管生成。K183位點的YY1突變消除了這些影響。p300的過表達增加YY1乳酸化并增強體外血管生成,并且p300抑制劑A485的給藥大大抑制了體內和體外的血管形成。該研究結果表明,小膠質細胞中YY1的乳酸化通過上調FGF2的表達在視網膜新生血管形成中發揮重要作用。靶向乳酸/p300/YY1乳酸化/FGF2軸可能為增殖性視網膜病變提供新的治療靶點。該研究于2023年4月發表于《Genome Biology》上,題為“YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2”,影響因子12.3。
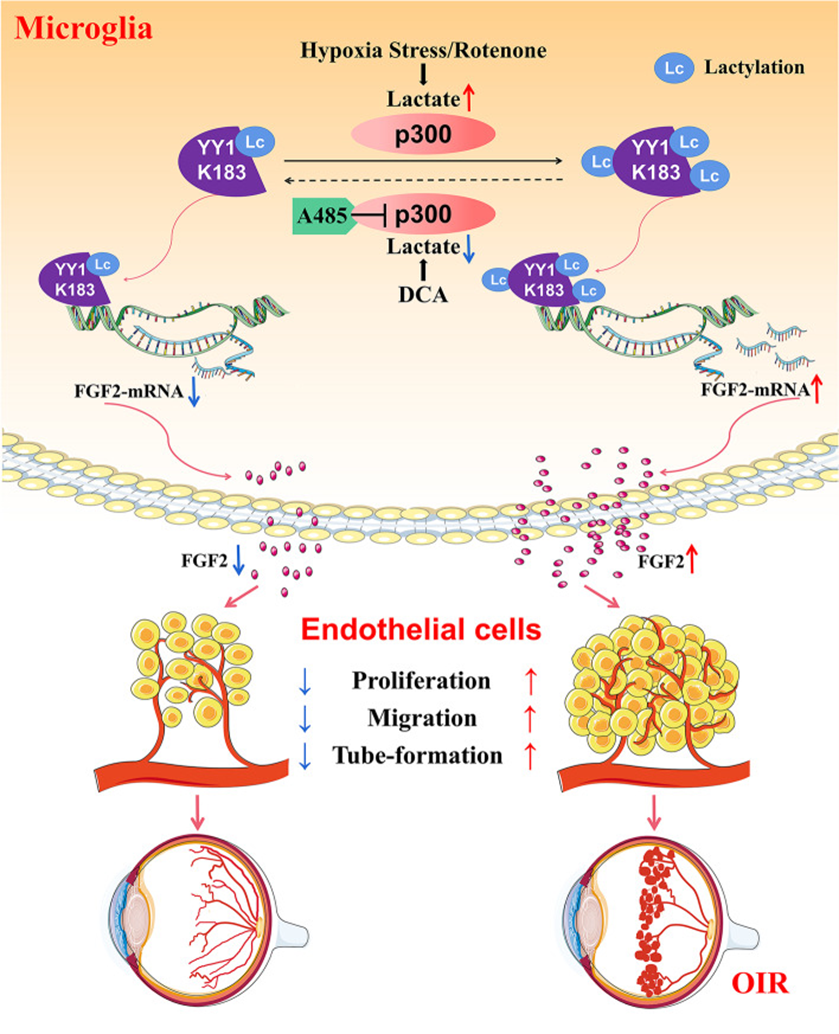
技術路線

研究思路
1. 視網膜小膠質細胞對視網膜新生血管形成至關重要
為了評估小膠質細胞在視網膜病變血管發病機制中的作用,開發了一種廣泛使用的OIR小鼠模型,該模型概括了人類增殖性視網膜病變,如ROP,如流程圖所示(圖1a)。建模后,視網膜小膠質細胞大量增殖并聚集在OIR視網膜新生血管形成部位周圍(圖1b)。為了評估小膠質細胞耗竭是否會影響OIR的進展,作者使用了CSF1R抑制劑(PLX3397),該抑制劑被證明可誘導明顯的小膠質細胞消融。加藥流程圖如圖1c所示(從P10到P17注射)。給藥后,作者觀察到病理性血管生成隨著小膠質細胞的耗竭而受到極大的抑制(圖1d)。這些結果證明了小膠質細胞在視網膜新生血管形成中的重要作用。
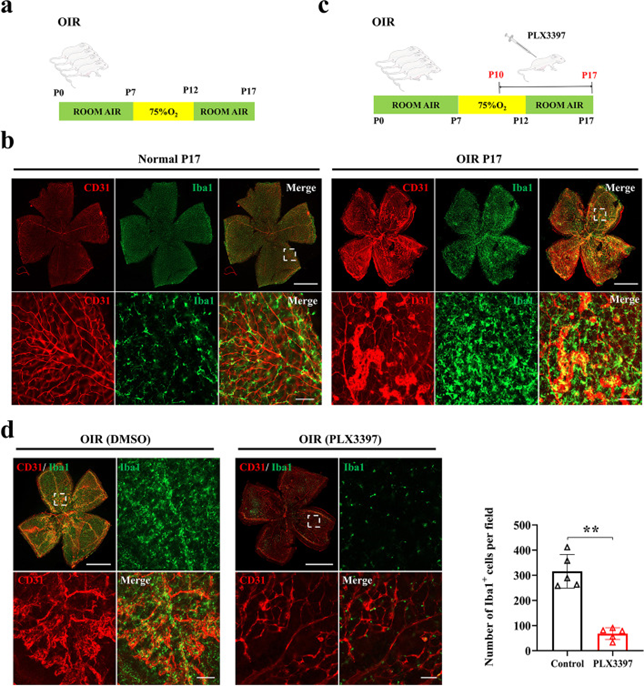
圖1 視網膜小膠質細胞對視網膜新生血管形成至關重要
2. 乳酸和乳酸化水平升高與視網膜新生血管形成有關
乳酸化來源于乳酸,參與多種細胞過程。為了確認乳酸和乳酸化是否參與缺氧誘導的增殖性視網膜病變,作者檢查了OIR小鼠的乳酸含量和泛賴氨酸乳酸化(Pan-Kla)水平。與年齡匹配的對照組相比,OIR小鼠P17(OIR新生血管形成的峰值)視網膜中的乳酸水平上調(圖2a),與臨床數據一致,與非ROP嬰兒相比,ROP嬰兒血液中的乳酸含量顯著增加(圖2b)。同時,與對照組相比,OIR組的Pan-Kla水平上調(圖2c)。然后,作者想知道小膠質細胞的乳酸化是否在視網膜血管疾病中起重要作用。OIR小鼠視網膜小膠質細胞中的乳酸化上調(圖2d)。值得注意的是,隨著小膠質細胞的耗竭,視網膜中Pan-Kla的水平相對下調(圖2e)。
為了研究乳酸化是否有助于視網膜病變的血管發病機制,作者應用了兩種化合物進行體內驗證實驗,即二氯乙酸鈉(DCA)和魚藤酮,它們已被證明可以通過調節乳酸的產生來調節乳酸化。DCA可以通過抑制丙酮酸脫氫酶激酶的活性來減少乳酸的產生,魚藤酮是線粒體呼吸鏈復合物的抑制劑,使細胞傾向于進行糖酵解并增加乳酸的含量。復方處理后,作者觀察到DCA組的乳酸和乳酸化水平降低,魚藤酮組的乳酸和乳酸化水平升高(圖2f,g),伴有DCA組視網膜新生血管的緩解和魚藤酮組新生血管的加重(圖2h)。這些結果表明乳酸化在視網膜新生血管形成中的重要作用。
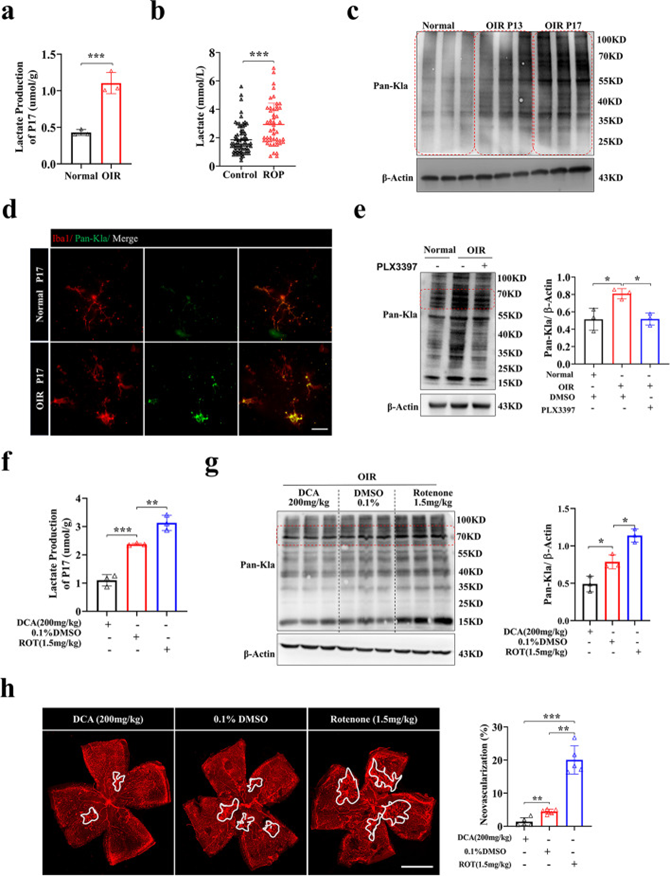
圖2 乳酸和乳酸化水平升高與視網膜新生血管形成有關
3. 小膠質細胞的高乳酸化促進體外血管生成
由于缺氧在血管生成過程中很重要,作者將人小膠質細胞克隆3(HMC3)細胞暴露于缺氧下,以探索乳酸化在小膠質細胞中的作用。隨著缺氧程度的增加,HMC3小膠質細胞的乳酸含量增加,乳酸化水平也升高,特別是對于55-70 kD范圍內的蛋白質(圖3a,b)。內皮細胞的增殖和遷移對于血管網絡的形成至關重要。為了探究乳酸化增加的小膠質細胞是否會影響內皮細胞的血管生成能力。作者將常氧或缺氧組的HMC3小膠質細胞與人視網膜微血管內皮細胞(HRMECs)共培養。隨著HMC3細胞缺氧時間的增加,HRMECs的管形成、球狀體萌芽、遷移和增殖的能力增強(圖3c-f)。為進一步證實小膠質細胞乳酸/乳酸化水平在血管生成發病機制中的重要作用,采用DCA和魚藤酮處理HMC3小膠質細胞,檢測乳酸/乳酸化水平。DCA組HMC3小膠質細胞的乳酸/乳酸化水平降低,魚藤酮組升高(圖3g,h)。然后,作者將DCA/DMSO/魚藤酮組的HMC3小膠質細胞與HRMECs共培養,以探索對HRMECs的影響。DCA組與HMC3細胞共培養的HRMECs的管形成、球狀芽、遷移和增殖能力減弱,魚藤酮組增強(圖3i-l)。這些結果表明,小膠質細胞中乳酸化升高是促進內皮細胞血管生成能力的原因。
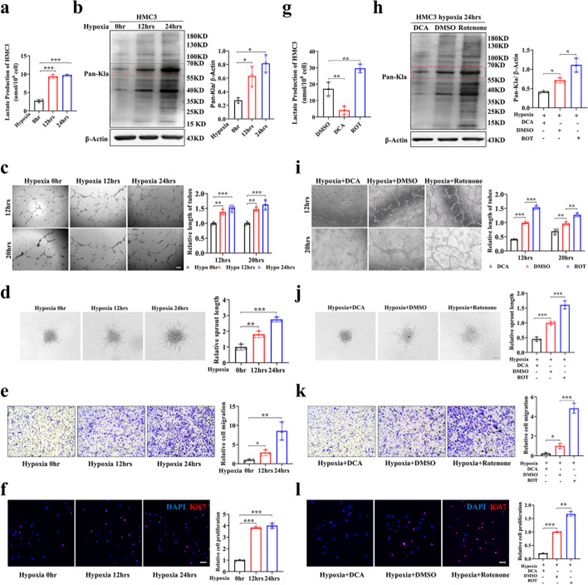
圖3 小膠質細胞的高乳酸化在體外促進血管生成
4. YY1在小膠質細胞中的乳酸化在調節血管生成中起著重要作用
為了表征缺氧下小膠質細胞中乳酸化的情況,作者用常氧或缺氧處理HMC3細胞24 h,然后通過4D無標記平臺方法進行乳酸組分析以鑒定差異乳酸化蛋白。對于乳酸組數據的詳細信息,共鑒定出6021個肽和3071個乳酸化肽。在HMC3細胞中總共鑒定了751個蛋白質中的3093個乳糖化位點,定位概率>0.75,FDR為1%。質量控制表明肽的分布在合理范圍。作者在缺氧條件下鑒定了乳酸化增加的67種蛋白質的77個位點和乳酸化降低的162種蛋白質的190個位點作為差異表達的乳酸化蛋白(DELPs)。乳酸化增加或減少的前20種蛋白質的相對定量如圖4a所示。對鑒定的DELPs行聚類分析以表征這些乳酸化蛋白的功能。分類分析表明,大多數DELPs在細胞核中發揮作用,并在調節DNA轉錄中具有潛在作用(圖4b)。使用相互作用基因/蛋白質檢索檢索工具(STRING)數據庫,DELPs的蛋白質相互作用網絡表明DELPs主要在結合過程中起作用,這與富集途徑相對應(圖4c)。
在這些DELPs中,YY1的乳酸化水平很大程度上上調(倍數變化3.56),而YY1是一種多功能轉錄因子,對血管生成有一定的調節作用。此外,觀察到的YY1條帶大小為68 kD,這與55-70 kD的范圍一致,根據作者之前的結果,該范圍主要由缺氧或用DCA和魚藤酮治療調節。作者在缺氧的HMC3小膠質細胞中觀察到YY1和Pan-Kla與雙標記免疫熒光的共定位。蛋白質組學分析在YY1(K183)中僅鑒定出一個乳酸化賴氨酸殘基,顯示出特征性的串聯質譜(MS/MS)譜圖,包括C端y離子和氨基末端b離子(圖4d)。為了驗證YY1乳酸化在缺氧條件下是否受到調控,作者使用專門的YY1-K183la抗體來檢測YY1乳酸化水平。與測序結果一致,YY1乳酸化在缺氧條件下顯著增加,YY1表達水平無差異(圖4e)。
接下來,作者將YY1的賴氨酸(K)183突變為精氨酸(R),其模擬蛋白質的脫乳酰化狀態,方法是用含有Flag標記的YY1WT或YY1K183R突變體的cDNA的慢病毒轉染HMC3小膠質細胞。Flag-YY1在WT和K183R突變體組中均過表達。在K183R突變體缺氧處理的HMC3小膠質細胞中,YY1乳酸化水平降低。與WT組相比,與K183R突變體HMC3小膠質細胞共培養的HRMECs顯示出減弱的管形成、球狀體萌芽、遷移和增殖能力(圖4f-i)。綜上所述,這些數據表明小膠質細胞中YY1的乳酸化起著血管生成的調節劑的作用。
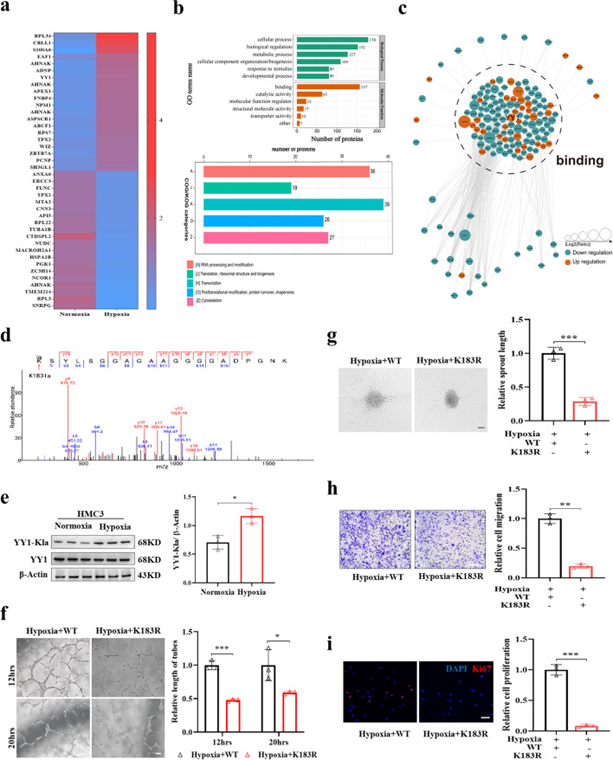
圖4 YY1在小膠質細胞中的乳酸化在調節血管生成中起著重要作用
5. YY1乳酸化通過調節FGF2表達促進血管生成
為了確定參與小膠質細胞介導的血管生成的潛在血管生成因子,作者在體外逆轉錄了從體內常氧、缺氧和小膠質細胞耗竭的視網膜和HMC3小膠質細胞中分離的mRNA,并比較了一些經典血管生成相關基因(VEGFA/FGF2/MMP9/MMP2/ANGPTL6)的表達水平。與對照組相比,OIR小鼠的FGF2和VEGFA mRNA表達水平顯著升高,而小膠質細胞耗竭后mRNA和蛋白水平僅FGF2水平顯著降低(圖5a,c)。同樣,缺氧在缺氧暴露后 12 小時內增加FGF2 mRNA 表達,并且在HMC3小膠質細胞中水平保持升高直到24 小時(圖5b,d)。FGF2在OIR視網膜小膠質細胞中的表達上調(圖5e)。與先前的研究一致,這些結果表明FGF2在小膠質細胞介導的新生血管形成中很重要。DCA和魚藤酮在體內和體外用于探討乳酸/乳酸化是否通過調節血管生成因子的表達來影響血管生成。在用DCA和魚藤酮處理的OIR模型中,DCA組FGF2的表達降低,但魚藤酮組在P17時增加。在暴露于DCA或魚藤酮缺氧的HMC3小膠質細胞中也發現了類似的結果。
YY1包含四個C2H2鋅指,用于結合位于許多啟動子和增強子中的特定DNA序列,促進或抑制轉錄。在YY1 K183R突變組中,FGF2在mRNA和蛋白水平上的表達均較低,而其他血管生成因子無差異(圖5f)。為了探究YY1乳酸化與FGF2表達的機制,作者檢索了Cistrome Data Browser數據庫,發現YY1可能直接促進FGF2的轉錄。作者在JASPAR網站上的FGF2啟動子中發現了三個預測的YY1結合位點,ChIP-qPCR顯示YY1可以結合FGF2轉錄起始位點上游的?1336至?1172bp區域。為了進一步探索缺氧條件下YY1的靶點,作者用YY1對這些血管生成因子的啟動子進行了ChIP-qPCR。值得注意的是,ChIP-qPCR分析顯示,缺氧促進了YY1與FGF2啟動子的結合,而不是其他血管生成因子(圖5g),主要是因為視網膜小膠質細胞不是分泌這種血管生成因子的主要細胞。K183R突變導致YY1與FGF2啟動子的結合受損,表明它受YY1乳酸化水平的調節(圖5h)。用雙熒光素酶報告基因系統驗證了YY1在缺氧條件下直接促進FGF2轉錄,YY1乳酸化位點突變導致轉錄能力降低的結果(圖5i)。同時,作者發現玻璃體內注射重組FGF2可逆轉小膠質細胞耗竭抑制的視網膜新生血管形。綜上所述,作者的研究結果表明,缺氧小膠質細胞釋放的FGF2受YY1乳酸化的調控,并在缺氧誘導的血管生成中起重要作用。
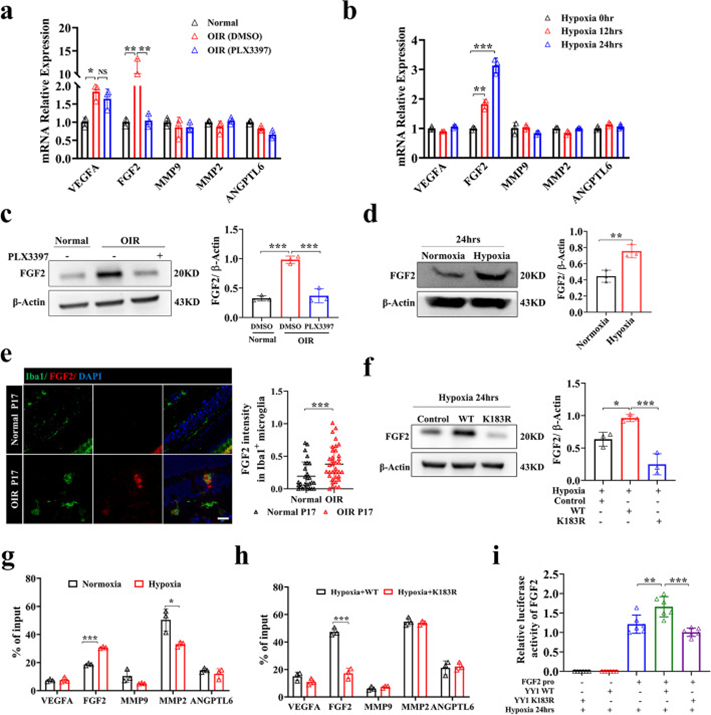
圖5 YY1乳酸化通過調節FGF2表達促進血管生成
6. p300影響YY1的乳酸化,進而調節體外血管生成
賴氨酸酰化是一系列普遍且進化上保守的PTM。乳酸化是一種新形式,可能與其他賴氨酸酰化類型共享相似的寫入器和橡皮擦。為了確定負責調節乳酸化水平的調節因子,作者在常氧和缺氧條件下檢測了已知酰化修飾寫入物(Tip60、p300和PCAF)和擦除劑(HDAC6和SIRT1)的表達模式。結果顯示,缺氧條件下HMC3小膠質細胞中p300、HDAC6和SIRT1水平顯著上調,Tip60水平略有上調(圖6a)。值得注意的是,根據Co-IP數據,只有p300與靶蛋白YY1結合,并且p300-YY1之間的相互作用隨著缺氧而增加(圖6b)。此外,作者還發現p300通過雙標記免疫熒光與YY1共定位(圖6c)。
為了進一步探討p300在調節乳酸化水平中的作用,作者在HMC3小膠質細胞中過表達p300,發現乳酸化水平顯著升高(圖6d)。有趣的是,p300的過表達增加了YY1的乳酸化水平,并伴有FGF2的上調(圖6e,f)。為了充分證實p300的作用,在隨后的實驗中使用了p300抑制劑A-485。A-485被證明是p300的CoA競爭性催化抑制劑,L-乳酰輔酶A是乳酸化所必需的。A-485處理后,YY1的乳酸化顯著降低,YY1轉錄調控的假設靶點FGF2的表達水平也下調(圖6g,h)。與p300過表達的HMC3小膠質細胞共培養的HRMECs的管形成、球狀芽、遷移和增殖能力增強,但與相應的對照組相比,A-485組減弱。
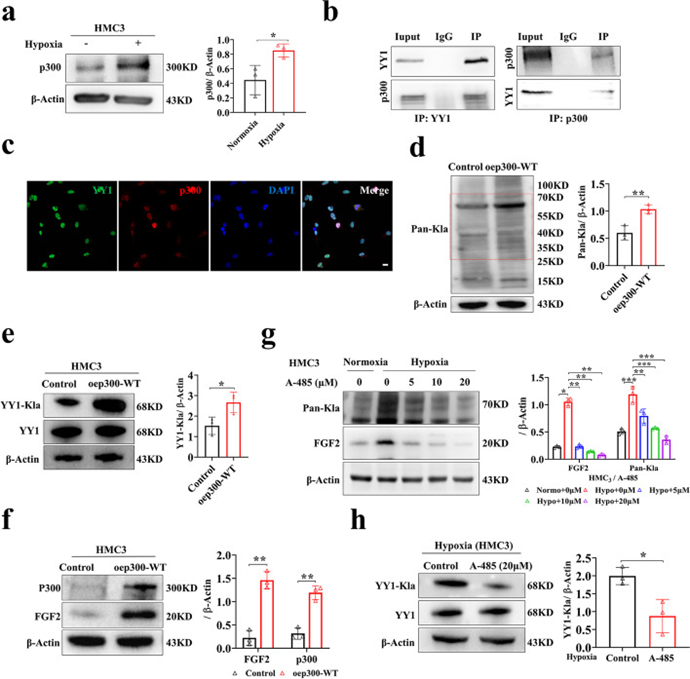
圖6 p300影響YY1的乳酸化,然后在體外調節血管生成
7. 抑制p300可減少視網膜小膠質細胞的YY1乳酸化并抑制OIR中的血管生成
了解了p300/YY1乳酸化/FGF2軸在小膠質細胞中的重要促血管生成作用,作者通過體內靶向p300和A485進一步評估了抗血管生成治療的療效。作者發現YY1乳酸化和p300的表達在OIR視網膜中上調(圖7a)。Iba1和YY1-K183la的雙重免疫熒光染色顯示OIR小鼠視網膜小膠質細胞中YY1乳酸化上調(圖7b)。在P14處進行玻璃體內給藥A485處理,服用A485后,Pan-Kla水平降低。有趣的是,A485處理降低了視網膜小膠質細胞中YY1的乳酸化(圖7c,d),伴有FGF2表達降低和視網膜新生血管緩解(圖7e,f)。小膠質細胞中乳酸化的減少可能與小膠質細胞活化減少有關(圖7g)。總體而言,這些結果表明,視網膜小膠質細胞中YY1乳酸化的上調與視網膜新生血管形成有關,表明阻斷p300-YY1乳酸化-FGF2信號傳導可能為治療視網膜新生血管疾病提供治療范式。
由于小鼠出生時沒有完整的視網膜血管網絡,并且視網膜處于相對缺氧狀態,作者想知道YY1乳酸化是否也在視網膜發育血管生成中發揮作用。作者發現YY1在視網膜發育的早期階段(出生后第5天)出現高乳酸化。YY1乳酸化水平在后期下降,表明YY1也可能在生理性血管生成中發揮作用。需要進一步的研究來深入探索它。
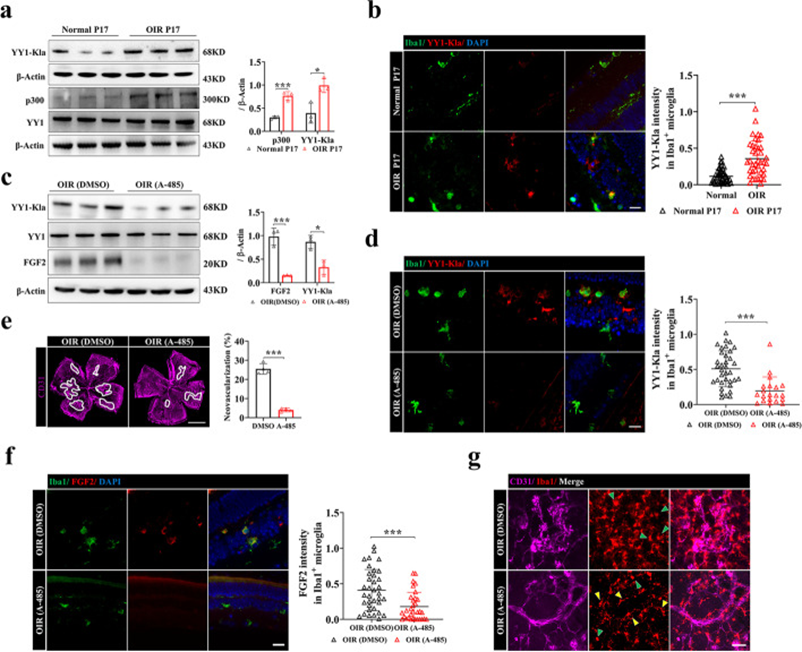
圖7 抑制p300可降低YY1的乳酸化并抑制OIR中的血管生成
實驗方法
小膠質細胞耗竭;動物建模與給藥;細胞培養;乳酸水平定量;免疫熒光染色;實時熒光定量PCR;WB;體外慢病毒感染和質粒轉染;基于泛抗體的翻譯后修飾富集;LC/MS分析和數據庫搜索;免疫共沉淀(Co-IP);ChIP檢測;雙熒光素酶報告實驗;HRMECs管形成實驗;基于球狀體的萌芽血管生成實驗;細胞遷移檢測;細胞增殖檢測;
參考文獻
Wang X, Fan W, Li N, Ma Y, Yao M, Wang G, He S, Li W, Tan J, Lu Q, Hou S. YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2. Genome Biol. 2023 Apr 21;24(1):87.