






新的鐵死亡增敏劑——地塞米松
地塞米松(Dexamethasone)被廣泛用于免疫抑制治療。本研究證明地塞米松對鐵死亡敏感,鐵死亡是一種鐵催化的壞死形式,以前被認為是導致急性腎損傷、心肌梗死和中風等疾病的原因,所有這些疾病都是由谷胱甘肽(GSH)耗竭引發的。本研究數據表明,地塞米松對鐵死亡的增敏是通過GR介導的DPEP1表達增加和GSH耗竭,具有臨床和治療意義。本研究于2022年2月發表在《Science Advances》IF 14.97期刊上。
技術路線:

主要實驗結果:
1、地塞米松對erastin誘導的鐵死亡敏感但對RSL3誘導的鐵死亡不敏感
作者通過5μM的erastin抑制system Xc-來誘導鐵死亡。通過流式細胞儀,檢測到在最初的30小時內,與只用erastin處理的組相比,erastin+地塞米松處理組的Annexin V/7AAD雙陰性細胞(活細胞)明顯減少,而Annexin V/7AAD雙陽性細胞的百分比明顯增加(圖1A和B)。為直接評估地塞米松對鐵死亡的影響,構建了一個3D打印的孵化室,它包含由玻璃片分隔的一個孔的兩個側面。該室允許我們在用對照或1μM地塞米松預孵化后30小時的整個觀察期內用一臺相機進行時間推移成像。將SYTOX綠和Annexin V添加到室的兩側,并用5μM的erastin刺激細胞。在地塞米松處理的細胞中檢測到SYTOX綠更早、更明顯的陽性反應(圖1C)。經過量化,在5μM erastin+1μM地塞米松處理后30小時,多達85%的細胞表現出SYTOX陽性信號,而在erastin刺激的對照組中大約只有40%的細胞是陽性的(圖1C)。總之,這些數據表明,1μM地塞米松能使HT1080細胞對erastin誘導的鐵死亡敏感。
接下來用小分子RSL3誘導鐵死亡,RSL3是谷胱甘肽過氧化物酶4(GPX4)的一種特征明確的抑制劑。與erastin誘導鐵死亡相反,RSL3誘導鐵死亡后,地塞米松預處理并沒有改變Annexin V/7AAD陰性細胞的百分比(圖1D)。erastin和RSL3誘導的鐵死亡之間的差異表明,地塞米松誘導的鐵死亡加速僅限于鐵死亡途徑的GSH調節部分。總之,這些數據表明,地塞米松不影響RSL3誘導的鐵死亡。
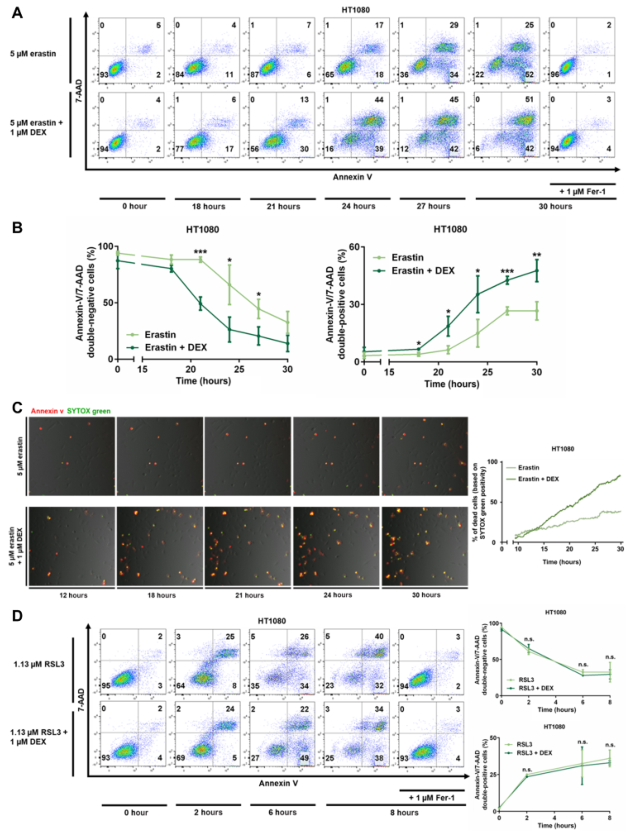
圖1 地塞米松對erastin誘導的鐵死亡敏感,但對RSL3誘導的鐵死亡不敏感
2、糖皮質激素,而不是醛固酮或脫氫表雄酮,對erastin誘導的鐵死亡敏感
鑒于類固醇激素之間的潛在冗余性,接下來詢問其他類固醇激素是否可能復制地塞米松對鐵死亡的影響。雖然地塞米松表現出最強的效果,但潑尼松龍和醛固酮(如果有的話,也只是非常小的程度)也在該試驗中表現出敏化作用,而與脫氫表雄酮(DHEA)共存則沒有效果(圖2A)。眾所周知,用糖皮質激素處理會導致GR受體下調,作者在用地塞米松和潑尼松龍刺激后的鐵死亡敏感細胞中證實了這種效應。相反,醛固酮和DHEA并沒有導致類似的GR的明顯下調。作為額外的對照,作者調查了礦物皮質激素受體(MR)的表達水平,已知它被醛固酮下調,發現地塞米松和潑尼松龍也下調了MR(圖2B)。總之,這些數據表明,地塞米松誘導的對鐵死亡的敏感性可能是通過GR介導的。
3、地塞米松誘導的對鐵死亡的致敏需要GR
地塞米松和其他糖皮質激素被描述為通過GR轉錄或以GR獨立的方式控制細胞過程。為測試GR在本系統中的作用,作者用Cas9穩定地轉染了HT1080細胞,并添加了引導RNA以產生HT1080 crKO(CRISPR敲除)的GR(圖2C)。與對照組細胞相比,GR的缺失完全逆轉了地塞米松或潑尼松龍介導的對erastin誘導的鐵死亡的敏感性(圖2D)。這些結果表明,糖皮質激素誘導的對erastin誘導的鐵死亡的敏感化需要GR。
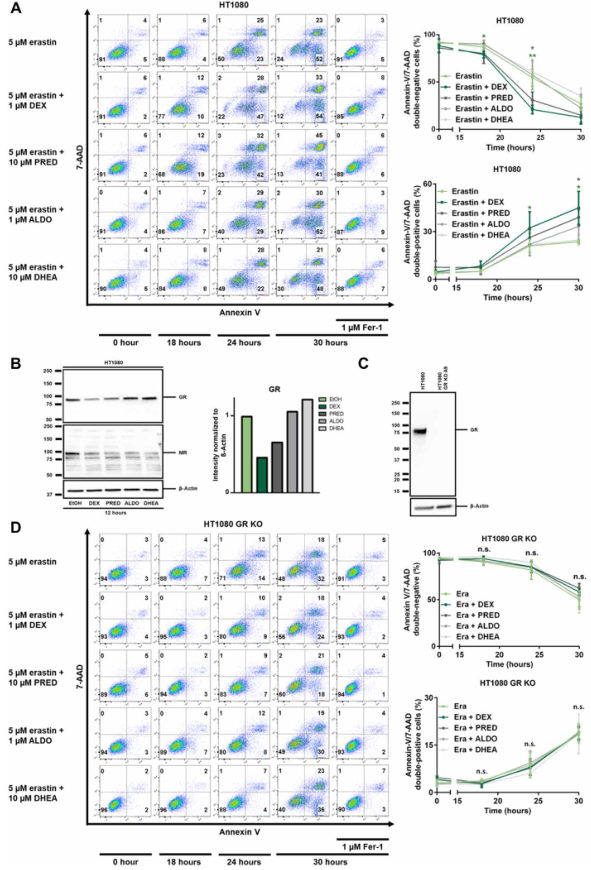
圖2 類固醇誘導的對erastin誘導的鐵死亡的致敏需要GR
4、地塞米松處理降低HT1080細胞的GSH
為了解地塞米松對GR的刺激是如何影響erastin誘導的而不是RSL3誘導的鐵死亡,首先觀察了鐵死亡途徑的已知標志物的蛋白表達水平。如圖3A所示,在地塞米松刺激后,檢測到ACSL4、SLC7A11、GPX4、TXNRD1、PRX1、TRX、CBS或CSE的表達沒有明顯變化。此外,檢測了HMOX1、GCLC和GCLM的蛋白表達水平(圖3B)。如預期的,HOMX1被erastin上調,而GCLC被地塞米松上調。為進一步了解細胞的氧化還原狀態,使用LC-ESI-QToF質譜來準確評估用1μM地塞米松處理后GSH的水平。如圖3C所示,作為GSH耗竭的對照,加入了erastin,雖然erastein完全耗盡了GSH,但地塞米松導致GSH含量減少50%以上,而沒有大量的細胞發生實際的鐵死亡(質膜破裂)。這個實驗解釋了為什么用地塞米松預處理能使erastin誘導的鐵死亡敏感,而RSL3誘導的鐵死亡沒有改變,因為后者繞過了鐵死亡誘導時對GSH耗竭的需要。
為確定GR激活下游的鐵死亡敏感性的潛在未知調節因子,進行RNA-seq。在最顯著的上調和下調的基因中,出現了幾個與氧化還原平衡有關的基因,如圖3D所示。在排名靠前的上調蛋白中,作者決定進一步驗證DPEP1。

圖3 對親代HT1080和GR-crKO細胞進行無偏倚的RNA測序,發現地塞米松誘導的基因與鐵死亡有關
5、敲低DPEP1逆轉地塞米松賦予的對erastin誘導的鐵死亡的敏感性
用1 μM地塞米松處理12小時后,DPEP1在蛋白水平上調(圖4A)。在Transwells培養的人原生腎小管上皮細胞中,DPEP1標記的免疫熒光在地塞米松處理后表現出增加,同時保持頂端表達/極性(圖4B)。總之,這些結果證實,地塞米松確實能上調DPEP1的蛋白表達。為測試DPEP1的功能作用,在HT1080細胞中敲除DPEP1(圖4C)。如圖4D所示,DPEP1的敲除逆轉了地塞米松對erastin誘導的鐵死亡的敏感性。

圖4 地塞米松介導的對erastin誘導的鐵死亡的致敏是由DPEP1介導的
6、地塞米松加速新分離的小鼠腎小管的LDH的自發釋放
文獻表明在新鮮分離的腎小管中,鐵死亡驅動自發的急性腎小管壞死。作者用這個體外模型來評估地塞米松對腎小管乳酸脫氫酶(LDH)釋放的影響。與對照處理的腎小管相比,地塞米松共處理導致LDH的釋放明顯增加,這種影響被Fer-1(圖5A和B)逆轉。地塞米松不能加速從DPEP1缺陷小鼠的小管中釋放LDH(圖5C和D)。與DPEP1在地塞米松誘導的敏化過程中的作用相一致,地塞米松/西司他丁共處理的腎小管與對照處理的腎小管相比,沒有表現出更高的LDH釋放水平(圖5E和F)。總之,這些數據表明,地塞米松在GR和DPEP1的介導下具有促進腎臟收縮的作用。

圖5 在野生型小鼠中,地塞米松加速新分離腎小管的鐵死亡,而在DPEP1基因敲除小鼠中則沒有
實驗方法:細胞培養和轉染,WB,流式細胞術,LDH釋放實驗,LC-ESI-QtoF質譜,免疫熒光,3D打印雙腔室同時活細胞成像,實時成像技術,RNA測序,小鼠模型和藥物處理,小鼠胸腺細胞和原代腎小管的分離。
參考文獻:
von M?ssenhausen A, Zamora Gonzalez N, Maremonti F, Belavgeni A, Tonnus W, Meyer C, Beer K, Hannani MT, Lau A, Peitzsch M, Hoppenz P, Locke S, Chavakis T, Kramann R, Muruve DA, Hugo C, Bornstein SR, Linkermann A. Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci Adv. 2022 Feb 4;8(5):eabl8920. doi: 10.1126/sciadv.abl8920. Epub 2022 Feb 2. PMID: 35108055; PMCID: PMC8809683.