






S6K1介導(dǎo)的PDK1磷酸化受損削弱AKT激酶活性和致癌作用
蛋白質(zhì)激酶作為細(xì)胞信號(hào)轉(zhuǎn)導(dǎo)的中心節(jié)點(diǎn),在時(shí)間和空間上能使下游底物發(fā)生磷酸化,以催化傳遞來(lái)自細(xì)胞外或細(xì)胞內(nèi)變化的信號(hào)。3-磷酸肌醇依賴(lài)蛋白激酶1 (PDK1)作為一種主激酶,在磷酸化和激活蛋白激酶A、B和C (AGC)家族激酶(包括AKT)中起著重要作用,但目前對(duì)PDK1的上游調(diào)控作用少有報(bào)道。本研究報(bào)道了核糖體蛋白S6激酶β1 (S6K1)直接磷酸化蛋白同源結(jié)構(gòu)域,并破壞PDK1與AKT的相互作用和活化。本文于2022年3月發(fā)表于 《Nature Communications》,IF=14.919。
本文技術(shù)路線(xiàn):

本文主要內(nèi)容:
1 S6K1直接磷酸化PDK1的Ser549位點(diǎn)
作者推測(cè)上游激酶可能介導(dǎo)PDK1-S549磷酸化,于是分析S549周?chē)被崆闆r,發(fā)現(xiàn)S549位點(diǎn)內(nèi)存在一個(gè)經(jīng)典的AGC激酶磷酸化共識(shí)基序位點(diǎn)(RxRxxS/T) (Fig 1a)。為了評(píng)估AGC激酶家族是否可以促進(jìn)PDK1在該位點(diǎn)的磷酸化,篩選了一組AGC激酶,包括AKT1 (myr-AKT1),AKT2 (myr-AKT2),S6K1 (S6K1- r3a)和SGK1 (SGK1-Δ60)。用內(nèi)源性的AKT底物磷酸化抗體檢測(cè)到S6K1,而不是其他的AGC激酶在內(nèi)源性水平上顯著增強(qiáng)PDK1磷酸化(Fig 1b)。胰島素誘導(dǎo)的PDK1磷酸化可被mTORC1或S6K抑制劑顯著拮抗,AKT抑制劑的拮抗程度較低,同時(shí)S6K下游底物核糖體蛋白S6的磷酸化事件減少(Fig 1c)。與此一致的是,S6K1可以顯著增強(qiáng)PDK1的磷酸化,且呈劑量依賴(lài)性(Fig 1d),而S6K1誘導(dǎo)的PDK1磷酸化可被mTORC1或S6K抑制劑減弱 (Fig 1e)。進(jìn)一步觀(guān)察發(fā)現(xiàn)PDK1磷酸化隨胰島素刺激而波動(dòng)。此外,胰島素在S6K1和S6K2雙敲除(S6K1 /2?/?)小鼠胚胎的成纖維細(xì)胞(mef)中極大促進(jìn)了PDK1磷酸化(Fig 1f)。提示S6K1在胰島素誘導(dǎo)中可能是必需的基因磷酸化。與這一發(fā)現(xiàn)一致的是,癌細(xì)胞中S6K1的缺失顯著減少了PDK1的磷酸化 (Fig 1g)。
為了驗(yàn)證S549是否確實(shí)是PDK1上主要的S6K1磷酸化殘基,作者發(fā)現(xiàn)S549A敲除降低了S6K1介導(dǎo)的PDK1磷酸化 (Fig 1h)。與這一發(fā)現(xiàn)一致的是,在體外S6K1可以直接磷酸化WT突變體,而不能磷酸化S549A突變體 (Fig 1i)。為了進(jìn)一步檢測(cè)S549位點(diǎn)的PDK1磷酸化,作者構(gòu)建并驗(yàn)證了磷酸化抗體特異性識(shí)別的PDK1-pS549,并觀(guān)察到S6K1確實(shí)可以在細(xì)胞的S549位點(diǎn)磷酸化PDK1 (Fig 1b-h),也能被mTORC1或S6K抑制劑可以抑制 (Fig 1c)。這些結(jié)果表明S6K1能直接磷酸化PDK1的Ser549殘基。
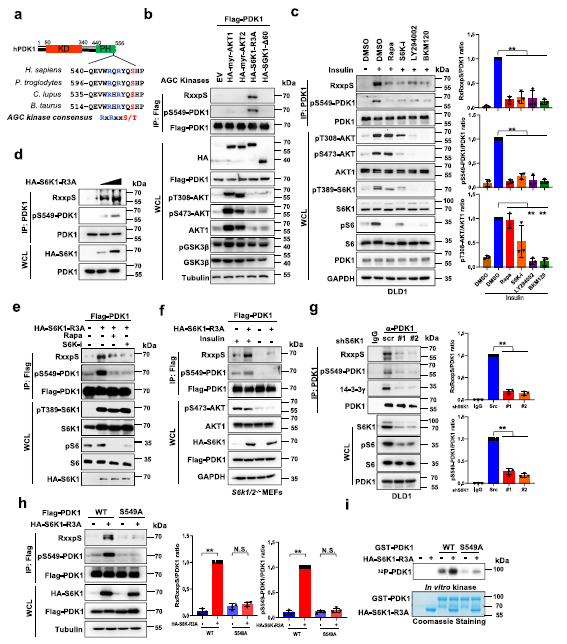
Fig1 S6K直接使PDK1在Ser549位點(diǎn)磷酸化
2 S6K1介導(dǎo)的PDK1磷酸化抑制AKT激酶活性和致癌功能
為了探究S6K1介導(dǎo)的PDK1磷酸化的生物學(xué)功能,作者將PDK1的不同突變形式(WT、S549A和S549D)引入DLD1-PDK1?/?細(xì)胞中,發(fā)現(xiàn)與WT或PDK1-S549D相比、PDK1-S549A表達(dá)顯著升高AKT-pT308 (Fig 2a)。此外,在胰島素處理?xiàng)l件下,如pGSK3β,與表達(dá)PDK1-WT-或S549D的細(xì)胞相比,DLD1-PDK1?/?表達(dá)PDK1-S549A的細(xì)胞中pT308-AKT及其下游底物顯著升高(Fig 2b,c)。
作者發(fā)現(xiàn),與WT或S549D突變體PDK1相比,引入缺磷型PDK1 (S549A)可以顯著促進(jìn)細(xì)胞集落形成和錨定獨(dú)立生長(zhǎng) (Fig 2d,e)。為了揭示PDK1-S549磷酸化在體內(nèi)對(duì)腫瘤生長(zhǎng)的作用,作者利用異種移植小鼠模型,發(fā)現(xiàn)與表達(dá)細(xì)胞S549的DLD1-PDK1?/?的細(xì)胞相比,PDK1-S549A的表達(dá)明顯促進(jìn)DLD1-PDK1?/?細(xì)胞腫瘤的生長(zhǎng) (Fig 2f–j)。同時(shí),與PDK1-WT或S549D的表達(dá)的腫瘤相比較,攜帶PDK1-S549A的腫瘤表現(xiàn)出更多的Ki67染色,同時(shí)AKT下游底物pS6增加 (Fig 2k,l)。這些結(jié)果表明,在體內(nèi)和體外,S6K介導(dǎo)的PDK1磷酸化負(fù)調(diào)控AKT激酶活性和致癌功能。
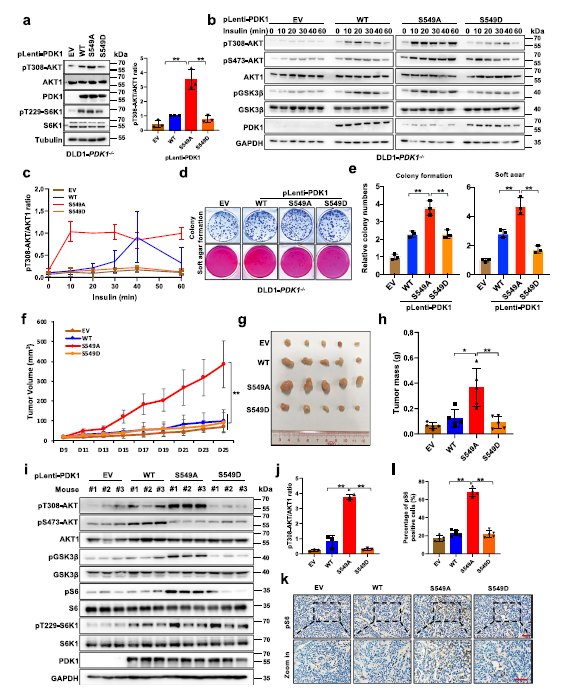
Fig2 PDK1的磷酸化抑制AKT激酶的活性和致癌功能
3 PDK1- S549磷酸化顯著減弱PDK1與PIP3的相互作用和質(zhì)膜定位
為了研究S6K1介導(dǎo)的PDK1磷酸化是否影響其激酶活性,作者對(duì)PDK1進(jìn)行了體外激酶測(cè)定。結(jié)果表明,S6K1-R3A的組成活性形式與PDK1- wt、S549A或S549D變體僅輕度影響PDK1磷酸化其底物AKT1的能力 (Fig 3a),表明S6K1介導(dǎo)的PDK1磷酸化對(duì)AKT激酶活性的抑制可能不是由于PDK1酶活性降低所致。接下來(lái),作者發(fā)現(xiàn)PDK1和S6K1-R3A可顯著降低PDK1與AKT的相互作用,且呈劑量依賴(lài)性,且S549處PDK1的磷酸化水平增加 (Fig 3b)。通過(guò)磷酸化IRS1/2來(lái)排除S6K已建立的負(fù)反饋?zhàn)饔玫挠绊懀^(guān)察PDK1-S549D或S549A與AKT之間的互作,結(jié)果發(fā)現(xiàn)PDK1-S549A的相互作用增強(qiáng),而S549D減弱,但不影響與其他PDK1底物(如SGK1、PLK1或S6K1)的相互作用 (Fig 3c)。
已有研究表明PDK1和AKT的相互作用主要發(fā)生在PIP3定位的質(zhì)膜上,PIP3的pull-down檢測(cè)結(jié)果顯示,S6K1-R3A顯著降低了WT的相互作用,而PDK1的S549A突變體與PIP3的相互作用沒(méi)有降低(Fig 3d)。同時(shí),抑制S6K1活性可以促進(jìn)PDK1與PIP3的相互作用(Fig 3e)。這些數(shù)據(jù)表明,S549磷酸化可以使PDK1從質(zhì)膜和PIP3中分離出來(lái),從而降低PDK1與AKT的相互作用。與這一發(fā)現(xiàn)一致,作者觀(guān)察到S6K1確實(shí)可以通過(guò)細(xì)胞分餾分析降低PDK1的膜定位或免疫染色 (Fig 3g)。因此,與WT或S549D形式的PDK1相比,S549A突變體增強(qiáng)了PDK1膜定位。
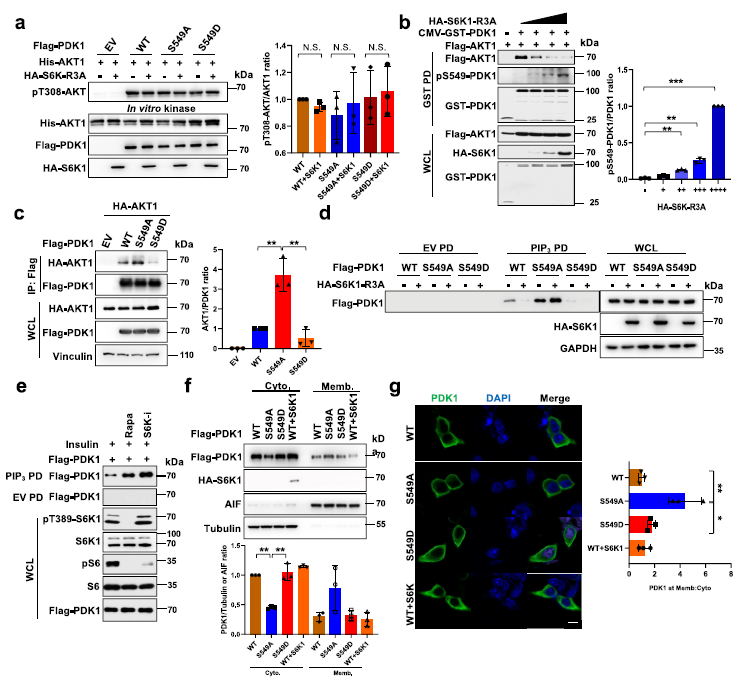
Fig 3 S6K1介導(dǎo)的PDK1磷酸化抑制PDK1膜定位和PIP3結(jié)合
4、14-3-3介導(dǎo)PDK1從PIP3中分離,并促進(jìn)其二聚
為了揭示磷酸化PDK1與PIP3解離并形成二聚體的潛在機(jī)制,作者假設(shè)存在能夠識(shí)別磷酸化PDK1的接頭蛋白,并進(jìn)一步干擾其與PIP3的相互作用。已有研究證實(shí)14-3-3接頭蛋白在蛋白室轉(zhuǎn)位中起主要作用,尤其是磷酸化蛋白。因此,作者對(duì)14-3-3家族成員進(jìn)行了篩選,發(fā)現(xiàn)14-3-3γ可以在外源和內(nèi)源水平上特異性地與PDK1相互作用 (Fig 4a–c)。值得注意的是,RxxpSxP這個(gè)基序與S6K1介導(dǎo)的PDK1磷酸化殘基(Ser549)重疊,并且在不同物種之間進(jìn)化保守 (Fig 4d)。有趣的是,與PDK1、AKT1、PLK1和SIN1衍生的不同PH結(jié)構(gòu)域相比,作者觀(guān)察到只有PDK1-PH參與14-3-3γ的結(jié)合。更重要的是,S6K1- r3a顯著增強(qiáng),而S6K1的消耗減少了PDK1與14-3-3γ之間的互作 (Fig 4g)。在細(xì)胞和體外實(shí)驗(yàn)中,S549D突變體PDK1增強(qiáng),而S549A突變體PDK1減弱PDK1與14-3-3γ的相互作用 (Fig 4e,f)。
先前有報(bào)道稱(chēng),磷酸化絲氨酸或蘇氨酸后的脯氨酸殘基對(duì)14-3-3相互作用至關(guān)重要。因此,無(wú)論在細(xì)胞內(nèi)還是體外,PDK1- p551a突變均能強(qiáng)烈減弱PDK1與14-3-3γ的相互作用。然而,P551A突變體并沒(méi)有消除S6K介導(dǎo)的PDK1與PIP3的分離 (Fig 4g),從而增加了AKT的相互作用,這不能被異位表達(dá)S6K1-R3A破壞(Fig 4g)。 S6K1-R3A或PDK1- S549d的異位表達(dá)同樣增強(qiáng)了14-3-3γ向PDK1的招募(Fig 4g)。此外,P551A突變體增強(qiáng)了其與PIP3的結(jié)合,S6K1-R3A的表達(dá)不會(huì)干擾PIP3的結(jié)合(Fig 4i)。
通過(guò)凝膠過(guò)濾的方法,作者觀(guān)察到PDK1和pS549-PDK1在其二聚體大小在150KD左右時(shí),與14-3-3γ共聚在相似的部分(Fig 4j),表明14-3-3γ可能是磷酸化PDK1進(jìn)行二聚反應(yīng)。此外,消耗14-3-3γ會(huì)破壞PDK1的二聚反應(yīng)(Fig 4k),并增加PDK1與AKT的相互作用。更重要的是,14-3-3γ敲除顯著提高了AKT及其下游靶點(diǎn)磷酸化水平(Fig 4l,m)。表明接頭蛋白14-3-3γ在S6K1介導(dǎo)的AKT被PDK1抑制過(guò)程中起重要作用。這些研究結(jié)果表明,14-3-3γ以S6K1介導(dǎo)的S549磷酸化依賴(lài)的方式與PDK1結(jié)合,使PDK1與PIP3分離,進(jìn)而促進(jìn)PDK1二聚化,從而抑制AKT激酶活性(Fig 4n)。
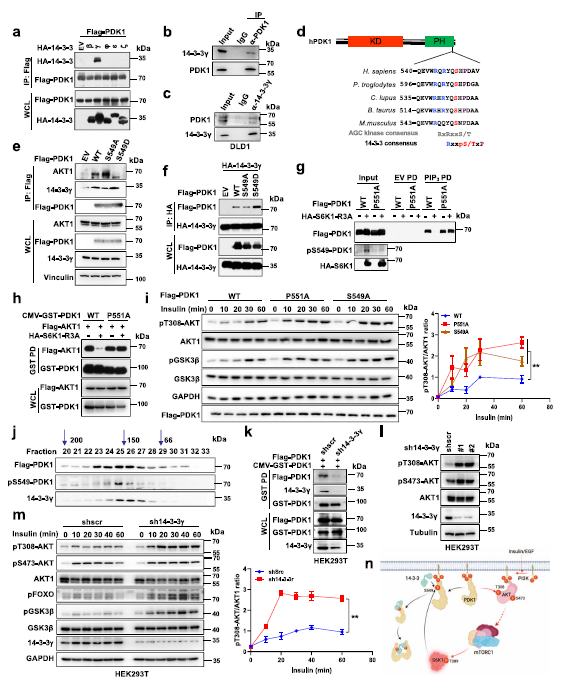
Fig4 14-3-3γ結(jié)合磷酸化的PDK1并將其從細(xì)胞質(zhì)膜上解離
5.患者相關(guān)的PDK1突變削弱其與14-3-3的相互作用,促進(jìn)AKT激酶活性和致癌功能
為探討S6K1介導(dǎo)PDK1磷酸化在腫瘤發(fā)生中的病理作用,對(duì)癌癥基因組數(shù)據(jù)進(jìn)行分析,結(jié)果發(fā)現(xiàn)患者相關(guān)的PDK1突變發(fā)生在S6K1介導(dǎo)的PDK1磷酸化或14-3-3γ結(jié)合區(qū)域附近 (Fig 5a)。
接下來(lái),這些突變被分成兩組,I型突變位于可能阻斷S6K1介導(dǎo)的磷酸化的區(qū)域; (R544K, R546P和S549N);II型突變(P551A, P551L和P551Q)可能在直接干擾對(duì)14-3-3γ的識(shí)別(Fig 5a)。結(jié)果發(fā)現(xiàn), I型突變以及程度較輕II型突變,顯著降低了S6K1介導(dǎo)的PDK1磷酸化(Fig 5b)。I型和II突變體顯著減弱PDK1/14-3-3γ相互作用(Fig 5c),從而增加了PDK1與PIP3的結(jié)合 (Fig 5d),以及它們的膜定位(Fig 5e–h)。值得注意的是,這些突變?cè)鰪?qiáng)了PDK1與AKT的相互作用(Fig 5i,j),同時(shí)增加了HEK293-sgPDK1細(xì)胞中AKT及其靶蛋白的磷酸化水平 (Fig 5k,l)。
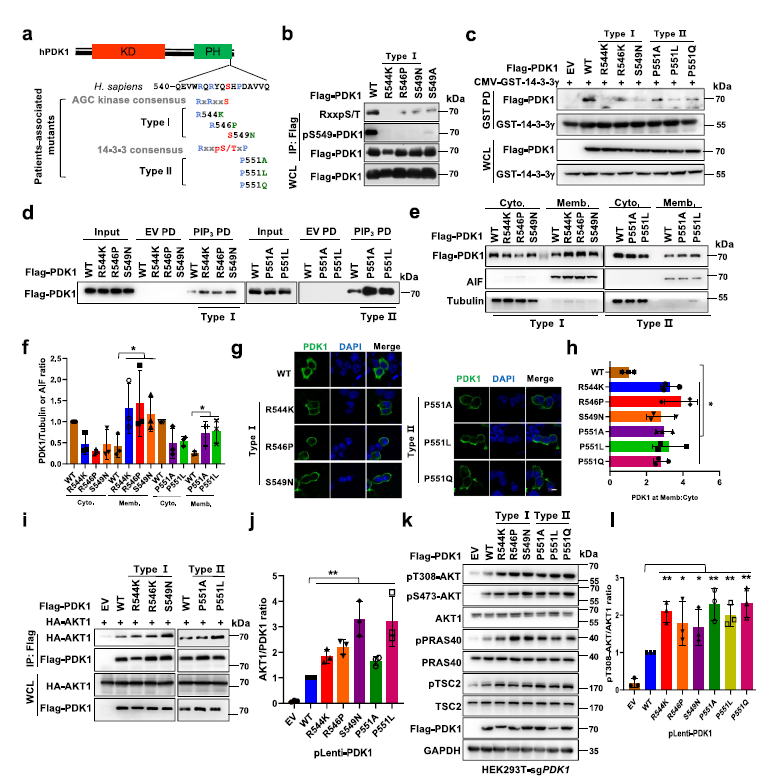
Fig5 患者相關(guān)的PDK1突變與PDK1膜位置和AKT激酶激活有關(guān)
此外,將這些變量重新引入DLD1-PDK1?/?或SW480-shP DK1細(xì)胞中增加了pT308-AKT的表達(dá) (Fig 6a),并促進(jìn)了癌細(xì)胞的致癌能力(Fig 6b,c)。此外,與表達(dá)WT-PDK1的細(xì)胞相比,這些突變體還促進(jìn)了異種移植小鼠模型中腫瘤的生長(zhǎng),并增加了AKT下游的pS6,以及增殖標(biāo)志物Ki67的表達(dá)(Fig 6d–g)。
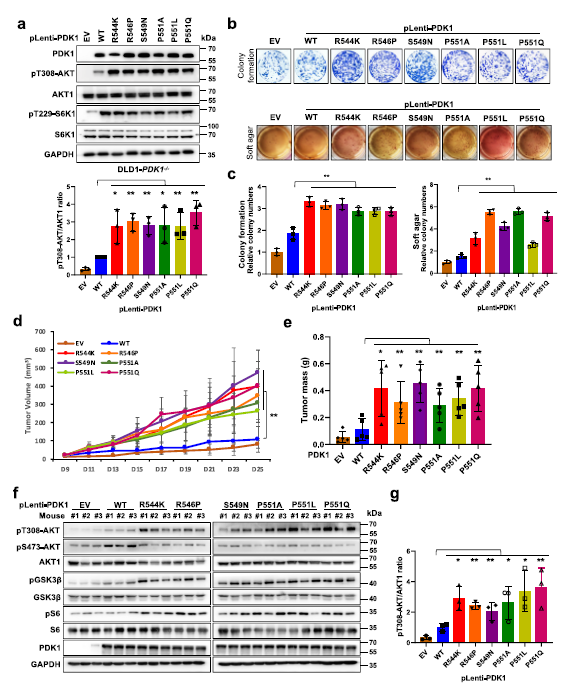
Fig 6患者來(lái)源的PDK1突變促進(jìn)AKT1的激活和致癌功能
總之,S6K1直接磷酸化PDK1的PH結(jié)構(gòu)域,增強(qiáng)PDK1結(jié)合14-3-3的能力,進(jìn)而解離PIP3和質(zhì)膜中的PDK1,導(dǎo)致PDK1/AKT相互作用減少,AKTT308磷酸化受損。這一觀(guān)察結(jié)果表明,S6K1以不同的負(fù)反饋方式和磷酸化依賴(lài)方式緊密控制AKT激酶的活性(Fig 7)。
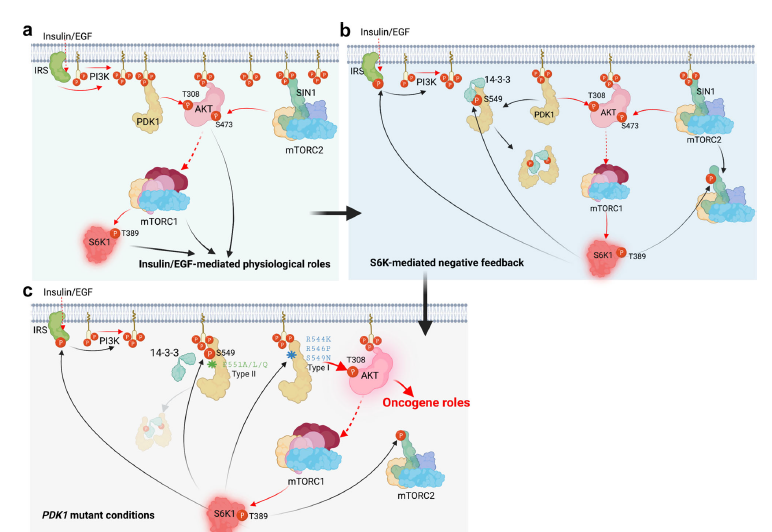
Fig 7 生理和病理?xiàng)l件下S6K1對(duì)PDK1負(fù)調(diào)控的模型
參考文獻(xiàn):
Jiang, Q., et al., S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat Commun, 2022. 13(1): p. 1548.