






SNRK對circ-SNRK的負反饋調節心肌梗死后心功能
心力衰竭(Heart failure,HF)作為一種慢性疾病威脅著人們的健康。作者認為預防心肌梗死后心肌細胞(CMs)丟失的研究可能會降低HF的發生率,從SNRK(蔗糖非發酵1相關激酶,可增加心臟線粒體效率)中鑒定出一個與HF相關的circRNA ,circ-SNRK,并闡明其調節機制。
技術路線:
主要結果:
采用左冠狀動脈前降支結扎法建立大鼠心衰模型,并通過超聲心動圖及心臟大體形態確保模型構建成功(Fig. 1A)。根據超聲心動圖結果將大鼠分為對照組、補償(Comp)組、失代償性(Decomp)組。術后4周(Ctrl + Comp組)及6周(Decomp組)采集心臟,樣品處理后進行高通量測序。在Ctrl vs Decomp和Comp vs Decomp分別篩選出923,1710個差異表達circRNA(fold change >2.0和p < 0.05)。此外,3組中存在507 個共有的circRNA,其中有498個circRNA在對照組和補償組表達豐度相似(Fig 1B)。將其進行GO及KEGG分析發現其中有43個circRNA參與了缺血相關通路(Fig 1C)。最后,通過FACS實驗檢測細胞凋亡,circ-SNRK在心肌細胞中表現出相對高的表達,并且與細胞凋亡密切相關(Fig. 1D)。
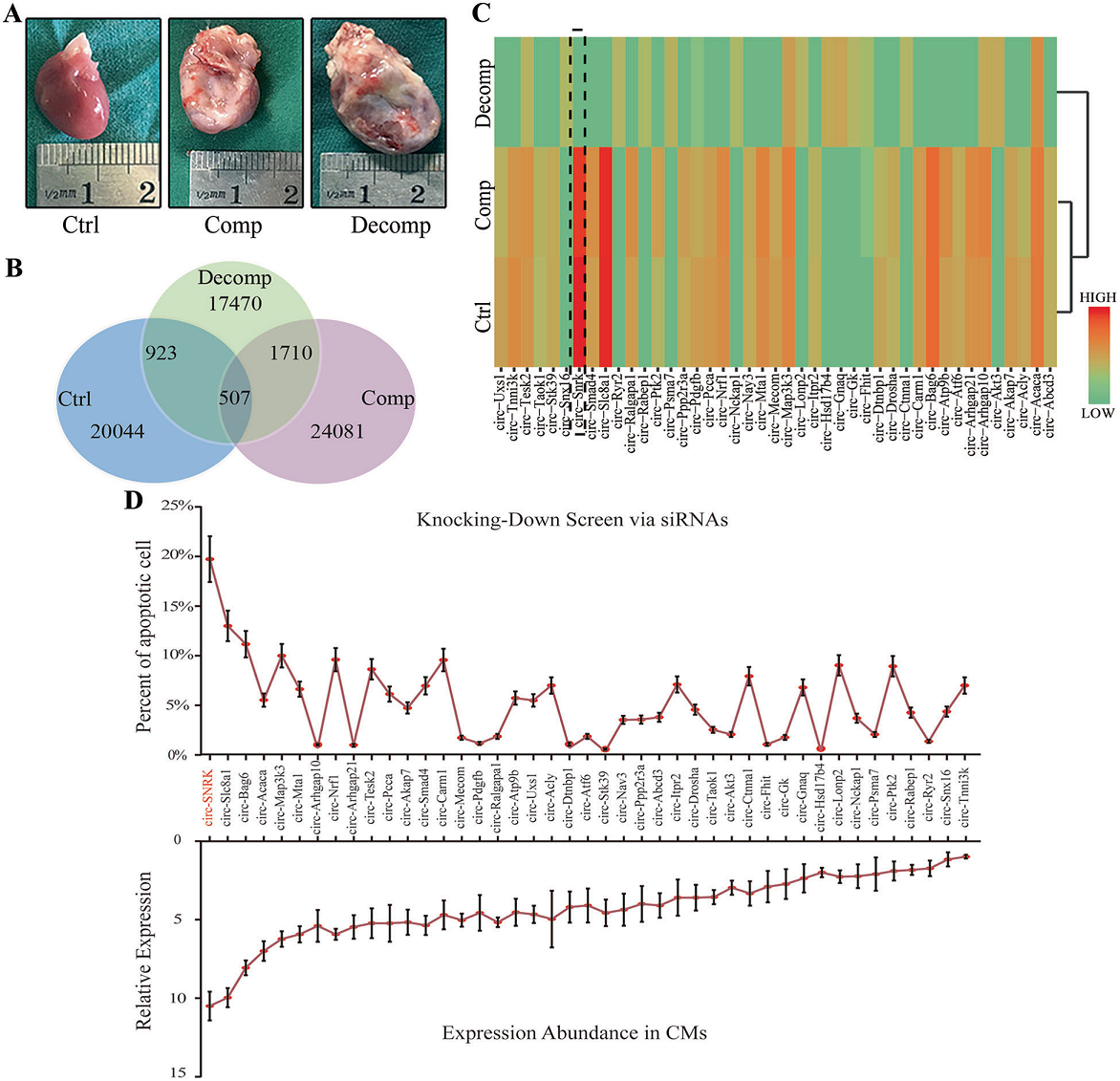
圖1 circ-SNRK的鑒定
2. circ-SNRK的特性及其對心肌細胞的影響
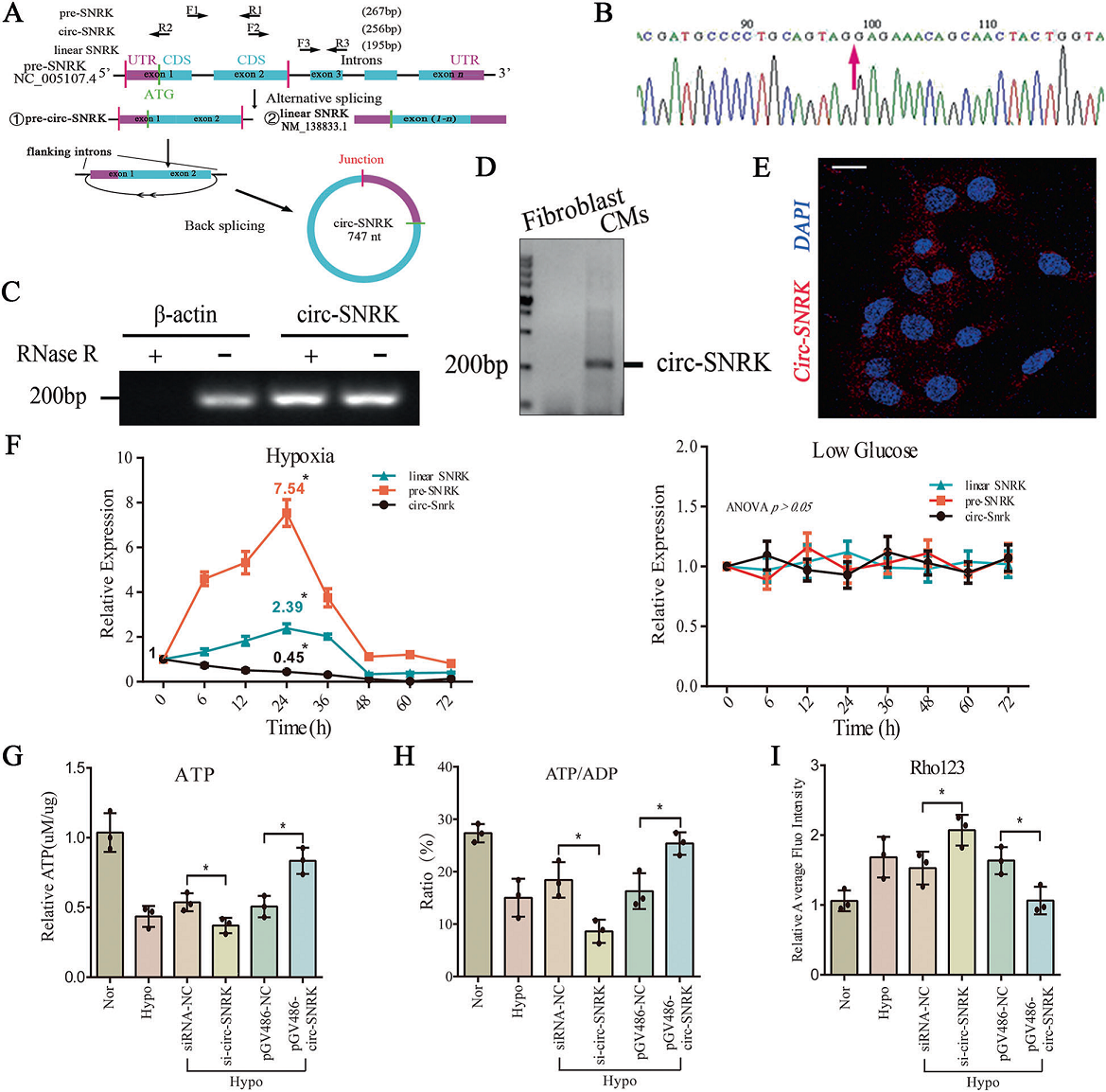
Sanger測序及RNase R實驗證實了circ-SNRK存在于心肌細胞中(Fig. 2A–C)。作者還發現,circ-SNRK在不同的器官中均有表達,在細胞質中有殘基,且不隨年齡變化(Fig. 2D, E)。qRT-PCR顯示,circ-SNRK在衰竭大鼠心臟中顯著降低,這與RNA-seq數據一致。心臟缺血對心肌細胞的主要影響是會造成氧、葡萄糖不足。我們的數據顯示在低氧處理下的乳鼠心肌小鼠的線性SNRK(mRNA)和pre-SNRK (precursor mRNA) 呈先上升后下降的趨勢,而circ-SNRK僅呈下降趨勢。而低血糖(<1 g/L)對其無明顯影響(Fig. 2A/F)。
為進一步研究其生理功能,作者分別用pGV486、siRNA來上調、下調circ-SNRK。Northern blot和RT-PCR結果顯示pGV486-circ-SNRK轉染HEK293細胞后可檢測circ-SNRK,只有circ-SNRK(而不是前SNRK或線性SNRK)被si-circ-SNRK下調。轉染后24 h,circ-SNRK 達到峰值(~25%),且其降解率低于線性SNRK。此外,我們發現circ-SNRK可以增加能量產生(Fig. 2G–I),能改善細胞凋亡,但對細胞自噬和焦亡無明顯作用。已有研究發現,circ-SNRK能通過SNRK蛋白作為能量調節器抑制細胞凋亡,于是作者主要關注其生理功能。
3. MiR-33通過SNRK介導circ-SNRK與ATP合成
之前在細胞質中發現circ-SNRK的殘基,暗示它可能通過其翻譯的多肽或作為miRNAs海綿發揮其功能。在發現它包含一個開放閱讀框后,作者將重構的帶有His標簽的circ-SNRK克隆到pGV486中。線性化ORF加上His標簽作為陽性控制。免疫印跡和免疫染色均未觀察到肽(~32 kDa),提示circ-SNRK在心肌細胞中不能轉化為肽。
接下來,作者推測miR-33可能是circ-SNRK的靶向的基因(Fig 3A)。雙熒光素酶報告基因和FISH檢測證實circ-SNRK可直接與miR-33結合(Fig. 3B)。同時,我們對AGO2進行RIP檢測,發現與對照組相比,circ-SNRK表達量明顯升高,進一步表明circ-SNRK可直接與miR-33 結合(Fig. 3C)。
接下來,作者發現miR-33可以降低心肌細胞中ATP和金屬蛋白酶(MMP)的合成,而miR-33抑制劑則有相反的作用(Fig. 3D)。此外,mut-circ-SNRK(沒有miR-33結合位點)對ATP合成和MMP的影響與pGV486-NC相似,而circ-SNRK的降低作用能被miR-33抑制劑挽救(Fig. 3E)。這些結果表明,miR-33介導circ-SNRK影響心肌細胞。
接下來,作者檢測了circ-SNRK是否通過SNRK調節能量代謝。結果發現,在小鼠中,circ-SNRK過表達會增加SNRK蛋白,circ-SNRK低表達會降低的SNRK蛋白的表達。同時, circ-SNRK引起的SNRK上調可以通過miR-33改善,而miR-33抑制劑可以挽救降低的SNRK。因此,circ-SNRK通過miR-33影響SNRK表達。然后,將缺氧處理的小鼠心肌細胞。通過轉染pGV486-circ- SNRC和si-SNRK或者si-circ-SNRK 和Ad-SNRK,作者發現,circ-SNRK過表達對ATP產生的影響被SNRK敲低所抑制,而circ-SNRK低表達的影響被SNRK過表達所回避(Fig. 3G)。說明circ-SNRK可以通過受miR-33 -SNRK軸,進而影響ATP合成。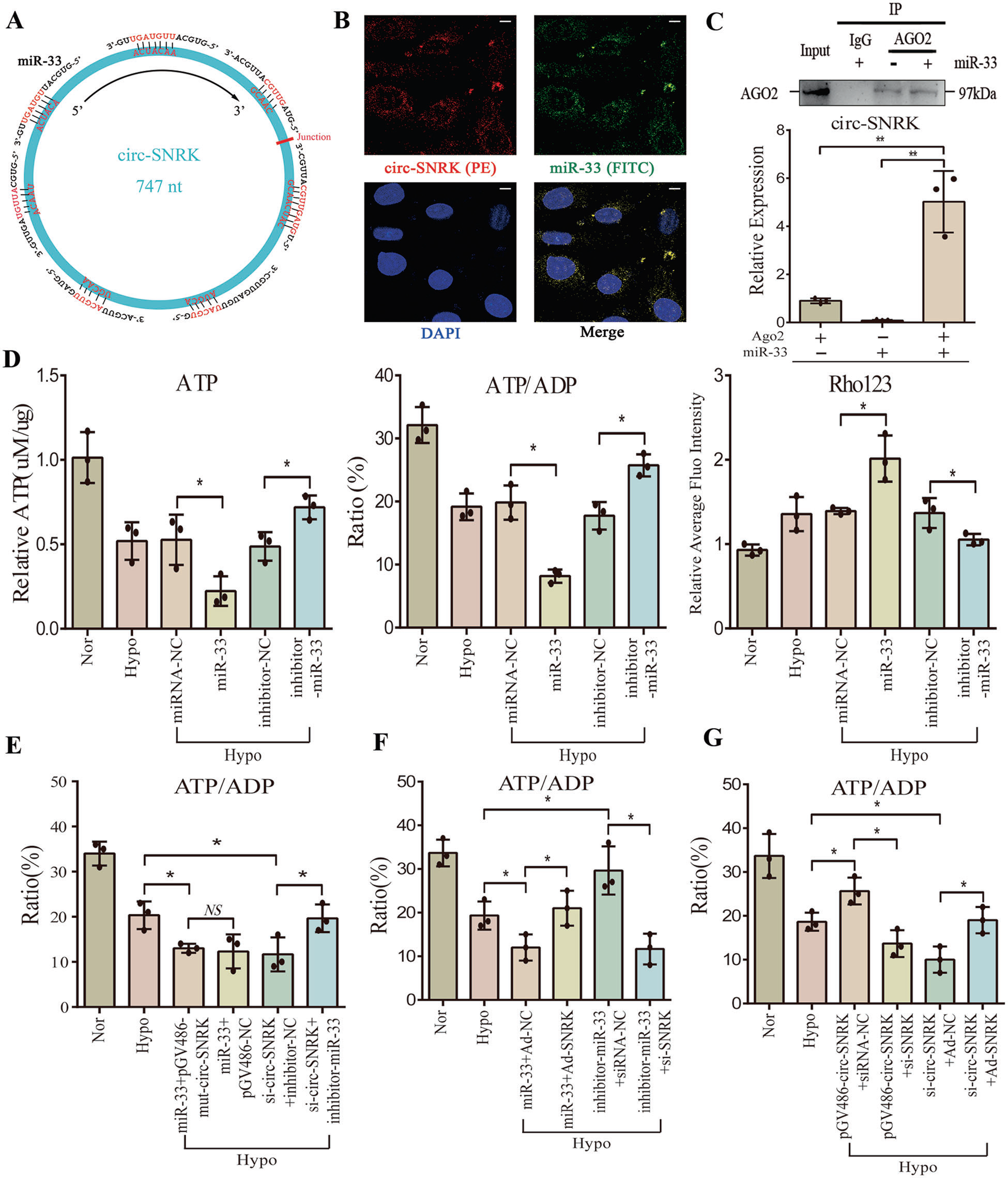
Fig3 MiR-33通過SNRK連接環狀SNRK與ATP合成
4. NOVA1促進circ-SNRK的形成,并直接與側翼內含子結合
鑒于miR-33可以提高circ-SNRK水平,而蛋白SNRK是miR-33的靶點,推測SNRK可能影響circ-SNRK的環化形成。qRT-PCR結果顯示,在心肌細胞中,circ-SNRK被si-SNRK上調,被Ad-SNRK下調,暗示SNRK在circ-SNRK的形成中起作用。
考慮到SNRK與circ-SNRK呈反向變化模式,它不應該直接連接到pre-circ-SNRK上,因為直接的相互作用會增加circ-SNRK的形成。為了識別連接SNRK和circ-SNRK的蛋白,通過側翼分析找到3個可能連接SNRK和circ-SNRK的RNA結合蛋白。進一步檢測,qRT-PCR結果顯示,只有si-NOVA1能降低Lnc-SNRK水平(圖4 b)。此外,我們檢測到在心肌細胞中過表達NOVA1會提高circ-SNRK表達水平 (圖4C)。以上結果表明,與SNRK蛋白不同,NOVA1可以促進circ-SNRK的形成。
通過分析pre-circ-SNRK的側翼內含子,作者在5 '內含子中發現了1個NOVA1的潛在結合位點,在3 '內含子中發現了3個NOVA1的潛在結合位點(Fig 4 d)。為了驗證相互作用,作者使用NOVA1的RIP,檢測了所有預測位點的序列(Fig 4E)。用生物素標記的RNA作為誘餌,檢測是否可以拉下NOVA1,結果所有位點均顯示陽性結果(Fig 4F)。表明NOVA1可以綁定到pre-circ-SNRK側翼,并對circ-SNRK有影響。
為了解NOVA1是否影響circ-SNRK的形成,作者將帶有NOVA1結合位點(wt或mut)的pre-circ-SNRK克隆到質粒(pCMV-wt-pre-circ-SNRK, pCMV-mut-pre-circ-SNRK)中,然后將其轉染到si-NOVA1處理過的HEK293中。兩種情況下均未觀察到circ-SNRK。然而,NOVA1過表達僅挽救了轉染了pCMV-wt-pre-circ-SNRK的HEK293的circ-SNRK,提示了NOVA1在circ-SNRK形成中的必要性(Fig. 4G)。
在PDB蛋白結構數據庫中觀察NOVA1結構,發現了NOVA1有超過兩個相似的RNA結合基序。此外,native PAGE的結果顯示,NOVA1可以在NRCMs中形成二聚體(圖4H)。于是推斷NOVA1具有促進circ-SNRK形成的能力。最后,作者發現蛋白NOVA1在缺氧狀態下沒有明顯變化,提示NOVA1在上述情況下能夠發揮其功能(Fig. 4I)。
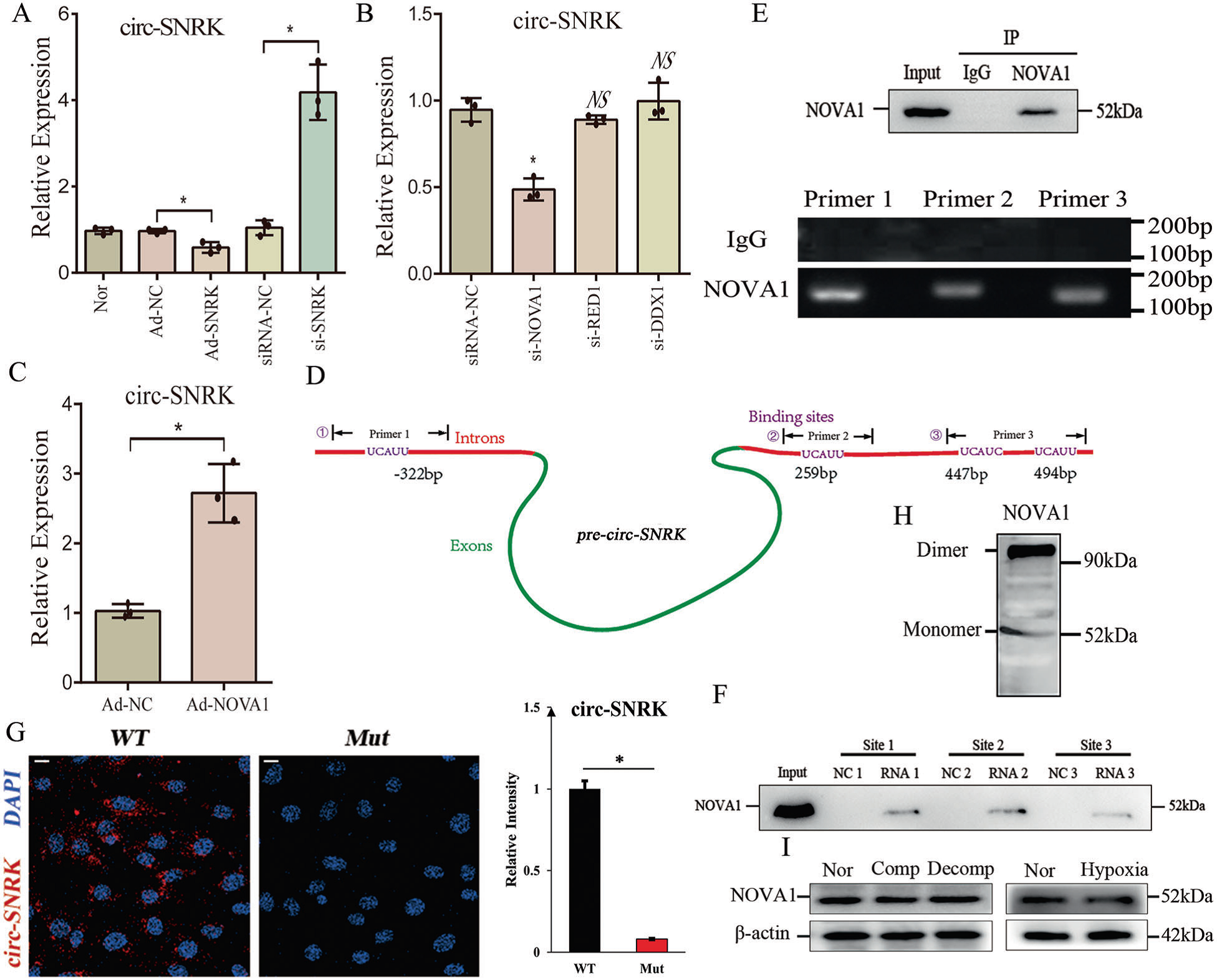
Fig. 4 NOVA1促進circ-SNRK的形成,并直接與側翼內含子結合
5. SNRK的55 kDa肽段能與NOVA1連接并影響circ-SNRK的形成
通過qRT-PCR實驗發現過表達NOVA1可以挽救circ-SNRK的下降,而Ad-SNRK的低表達則有相反的作用,提示SNRK通過NOVA1影響circ-SNRK的形成(Fig 5A)。進一步研究發現NOVA1 mRNA不受SNRK蛋白的影響(Fig 5B)。接下來進行SNRK的免疫共沉淀實驗,發現NOVA1被下拉(Fig 5C),NOVA1的下拉肽分子量為55 kDa,可被SNRK抗體所識別,說明它是SNRK蛋白的一部分(Fig 5D)。構建pCMV-N-His-SNRK和pCMV-SNRK-C-His探索其肽端,通過液相色譜串聯質譜分別發現了約30 kDa和55 kDa肽段(Fig 5E),進一步分析SNRK的結構(Fig 5F),最終發現這個55kDa的肽段屬于SNRK的C端。
已有研究表明,SNRK與細胞死亡有關,兩個結構域的連接包含Asp殘基(Caspases的底物),這都表明SNRK可能被Caspases降解。用Z-vad (Caspases的全局抑制劑)培養NRCMs 48小時,發現其55kda肽顯著下調(Fig 5G),進一步利用SNRK作為誘餌來鑒定哪些Caspase (Caspase 3/6/7)在這里發揮作用,發現只有Caspase 3參與其中和SNRK結合(Fig. 5H)。最后,作者通過 si-Caspase 3降低了NRCMs中的Caspase 3,發現55kda肽水平隨時間的增加而降低;然而,整體SNRK增加(圖5I)。綜上所述,SNRK可以被激活的Caspase 3降解分為兩個肽段,其中的55kda肽段直接與NOVA1相互作用。
此外,在Z-vad處理后,將pCMV-SNRK或pCMV-55kDa轉染新生大鼠心肌細胞48小時,探索55kDa肽能否影響circ-SNRK的形成,qRT-PCR顯示circ-SNRK僅受55 kDa肽影響(Fig 5J)。免疫熒光實驗顯示C端肽位于細胞質和細胞核中,而N端肽僅殘基在細胞質中,表明只有55 kDa肽能進入細胞核(Fig 5K)。研究SNRK如何調節NOVA1對circ-SNRK形成的影響,用放線菌素D處理在NACM中上調或下調55 kDa肽,發現NOVA1蛋白沒有明顯變化,提示55 kDa肽對NOVA1的穩定性沒有影響(圖5L)。轉染和qRT-PCR實驗推測NOVA1與55 kDa肽的相互作用可以抑制NOVA1與內含子的結合(Fig 5M)。綜上實驗表明SNRK通過NOVA1影響circ-SNRK的形成。

圖5 SNRK的55 kDa肽段能與NOVA1連接并影響環狀SNRK的形成
6. 缺氧通過NF-κB通路增加SNRK的轉錄
低氧誘導了心肌細胞內SNRK、線性SNRK和circ-SNRK 的變化。為了闡明其機制,于是作者對缺氧相關轉錄因子進行分析,包括HIF(低氧誘導因子)、NF-κB(核因子NF-kappa- b)或AP-1(激活蛋白-1)。分別通過抑制劑進行處理,發現僅在JSH-23處理下,缺氧條件下的心肌細胞pre-SNRK顯著降低(Fig. 6A)。此外,生物信息分析提示在SNRK的啟動子中NF-κB有三個可能的結合位點(Fig. 6B)。因此,作者推測缺氧可能通過NF-κB通路影響SNRK轉錄。然后,利用NF-κB復合體中最豐富的元素pCMV-P65上調P65水平。
免疫熒光顯示在pCMV-P65轉染的新生大鼠心肌細胞中,P65的核表達增加(Fig.6C)。新生大鼠心肌細胞中pre-SNRK的顯著增加表明NF-κB復合物可以激活SNRK轉錄(Fig 6D);在si-P65轉染的新生大鼠心肌細胞中pre-SNRK的減少進一步支持了這一觀點(Fig 6E)。接下來,作者針對預測位點在P65啟動子中設計了三種引物 (Fig 6B)。采用染色質免疫沉淀法檢測P65的相互作用。結果發現可以檢測到第二個位點的序列(Fig 6F)。作者進一步將SNRK的wt或mut啟動子克隆到pGL4.27載體中,顯示心肌細胞中共轉染pCMV-P65和pGL 4.27-wt-SNRK啟動子的熒光強度明顯高于對照組(Fig. 6G)。最后,發現在缺氧治療的心肌細胞或心肌梗死后P65升高,表明pre-SNRK過表達與心臟中的P65相關(Fig.6 h)。綜上所述,缺氧可以通過NF-κB通路激活SNRK轉錄。
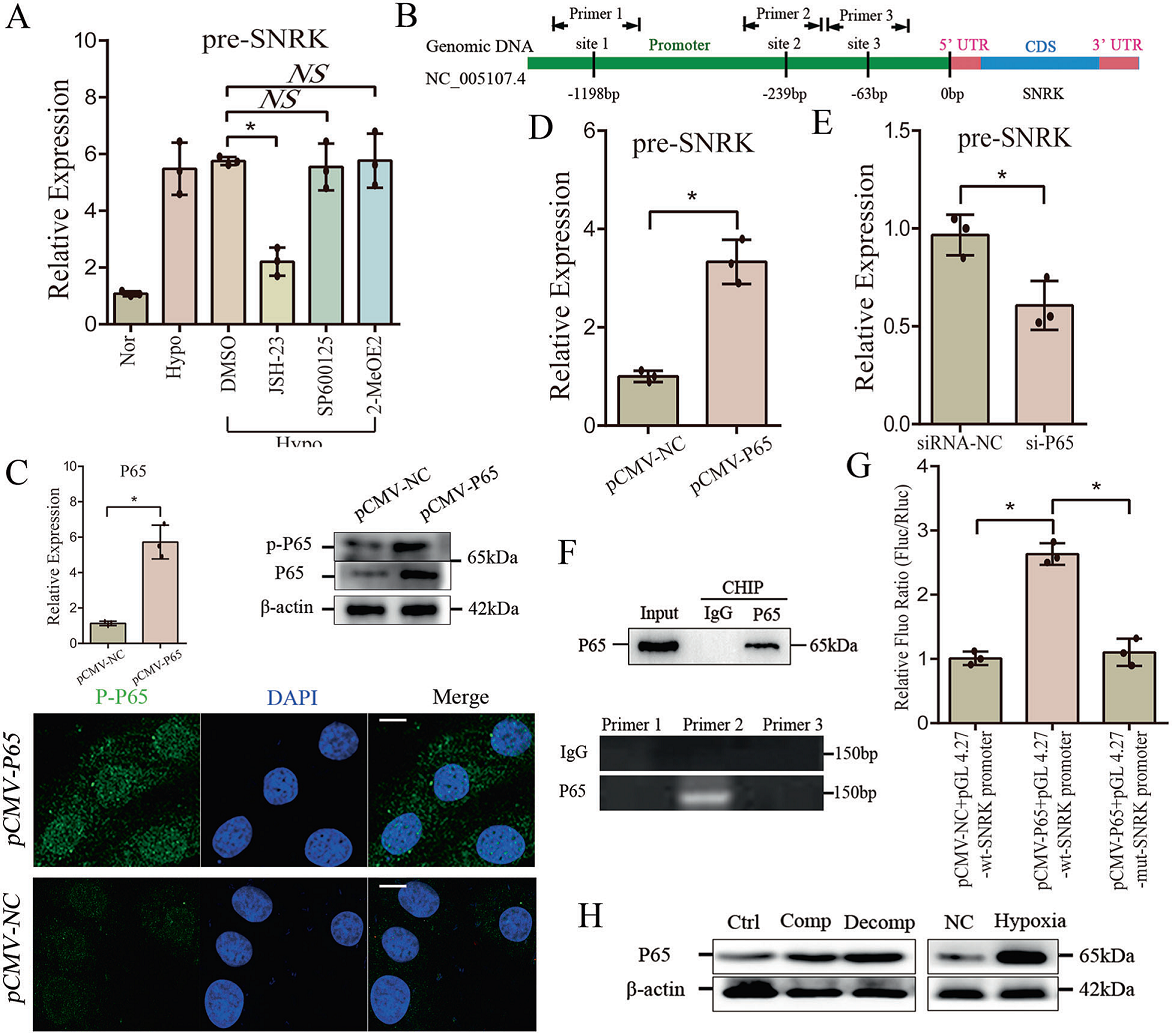
圖6 缺氧通過NF-κB通路促進SNRK的轉錄
7. Circ-SNRK改善心肌梗死大鼠心功能
作者接下來研究了circ-SNRK是否能在體內改善心肌梗死后心功能。通過病毒載體(AAV9)建立了過表達(MI-circ-SNRK 組)或不表達circ-SNRK (MI對照組)的HF大鼠模型。qRT-PCR結果顯示MI-circ-SNRK組circ-SNRK顯著上調(Fig 7A)。三色染色顯示SNRK組心肌梗死面積顯著降低(Fig 7B)。MI-circ-SNRK組超聲心動圖指標(LVEF、FS、LVPW)均較高,提示circ-SNRK過表達大鼠心功能較好(Fig 7C, D)。MI-circ-SNRK組ATP和ATP/ADP比值的升高表明其在能量代謝中的有益作用(Fig 7E,F)。Caspase 3/Parp-1免疫印跡和Tunel實驗表明circ-SNRK可以改善細胞在體內的死亡(Fig 7G-H)。研究circ-SNRK在體內是否通過SNRK發揮其功能,研究數據顯示SNRK和相關蛋白的表達與預期一致且不同組間miR-33無變化,這些與體外實驗結果一致(Fig 7I-J)。這些結果表明circ-SNRK可以通過miR-33 -SNRK軸保護心臟免于能量耗盡,進而改善心肌梗死后的心功能。

Fig. 7 Circ-SNRK可改善大鼠心肌梗死后心功能
circ-SNRK與SNRK的負循環調節了能量代謝,可能是治療心力衰竭的一個潛在靶點。
參考文獻: Wang ZY, Liu XX, Deng YF. Negative feedback of SNRK to circ-SNRK regulates cardiac function post-myocardial infarction. Cell Death Differ 2021.